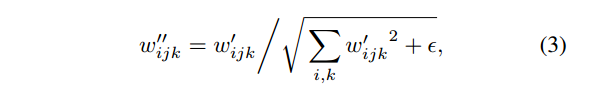
这个系列从2022年开始,一直更新使用R语言分析数据及绘制精美图形。小杜的生信笔记主要分享小杜学习日常!如果,你对此感兴趣可以加入该系列的学习。
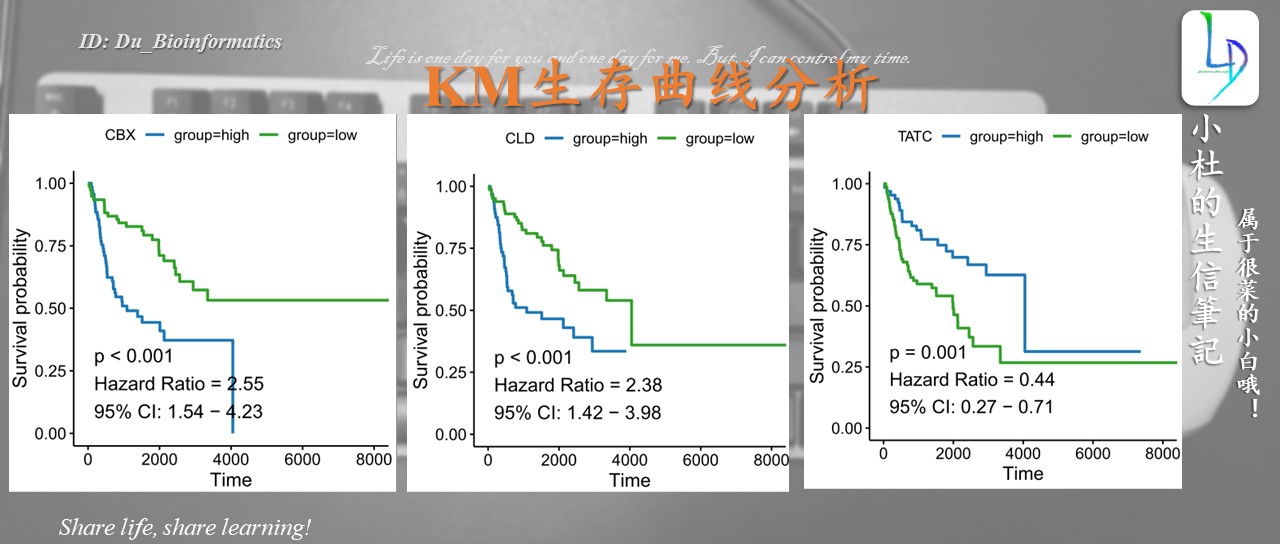
最终图形

本期图形代码
##'@KM生存分析
##'@20230807
##导入包
library(survival)
library(survminer)
##导入数据
setwd("E:\\小杜的生信筆記\\2023\\20230807-KM生存分析")
##
svdata <- read.csv("Input_data.csv",header = T,check.names=F,row.names = 1)
> svdata[1:10,1:5]
futime fustat CBX CLD CADL
sample01 2450 1 10.64 7.31 10.18
sample02 7367 0 9.38 7.43 7.16
sample03 8437 0 9.36 8.56 8.53
sample04 807 1 9.42 7.80 7.70
sample05 3343 1 10.20 7.28 9.52
sample06 459 1 10.34 11.00 9.67
sample07 556 1 9.63 10.65 9.12
sample08 3898 0 15.00 10.70 14.38
sample09 377 1 12.79 10.28 12.23
sample10 3869 0 11.40 10.93 9.78
数据过滤
# 找best separation用的是survminer的函数,避免无法切分数据集的情况,添加了4和5
pct = 0.1
## 1. 提取表达谱
expr <- svdata[,3:ncol(svdata)] ## 第三列以后的数据
head(expr)
# # 3. 取方差大于1的基因
expr <- expr[,apply(expr, 2, sd) > 0]
# 4. (关键) 取表达量为0的样本数目小于总样本数pct%的样本,这里pct默认为0.1,即10%
expr <- expr[,apply(expr, 2, function(x) sum(x == 0) < pct * nrow(expr))]
# 5. (关键) 基因名出错会导致出现只有一个组的情况
colnames(expr) <- make.names(colnames(expr))
# 6. 数据合并
svdata <- cbind.data.frame(svdata[,1:2],expr)
bestSeparation统计
res.cut <- surv_cutpoint(svdata,
time = "futime",
event = "fustat",
variables = names(svdata)[3:ncol(svdata)],
minprop = 0.3) #默认组内sample不能低于30%
高低组分类
res.cat <- surv_categorize(res.cut)
my.surv <- Surv(res.cat$futime, res.cat$fustat)
统计结果
# 建一个空的dataframe,用来存放统计量
ptable <- data.frame(matrix(NA,ncol(svdata)-2,5))
colnames(ptable) <- c("ID", "pvalue", "HR", "CIlow", "CIup")
批量绘图
for (i in colnames(res.cat)[3:ncol(svdata)]) {
#i = "ATAD3A"
group <- res.cat[,i]
survival_dat <- data.frame(group = group)
fit <- survfit(my.surv ~ group)
##计算HR以及95%CI
##修改分组参照
group <- factor(group, levels = c("low", "high"))
data.survdiff <- survdiff(my.surv ~ group)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
HR = (data.survdiff$obs[2]/data.survdiff$exp[2])/(data.survdiff$obs[1]/data.survdiff$exp[1])
up95 = exp(log(HR) + qnorm(0.975)*sqrt(1/data.survdiff$exp[2]+1/data.survdiff$exp[1]))
low95 = exp(log(HR) - qnorm(0.975)*sqrt(1/data.survdiff$exp[2]+1/data.survdiff$exp[1]))
if (p.val > 0.05) next
n = n + 1
ptable$ID[n] <- i
ptable$pvalue[n] <- p.val
ptable$HR[n] <- HR
ptable$CIlow[n] <- low95
ptable$CIup[n] <- up95
HR <- paste("Hazard Ratio = ", round(HR,2), sep = "")
CI <- paste("95% CI: ", paste(round(low95,2), round(up95,2), sep = " - "), sep = "")
#按照基因表达量从低到高排序,便于取出分界表达量
svsort <- svdata[order(svdata[,i]),]
# # 传统生存曲线
pl[[i]]<-ggsurvplot(fit, data = survival_dat ,
#ggtheme = theme_bw(), #想要网格就运行这行
conf.int = F, #不画置信区间,想画置信区间就把F改成T
#conf.int.style = "step",#置信区间的类型,还可改为ribbon
censor = F, #不显示观察值所在的位置
palette = c("#1f78b4","#33a02c"), #线的颜色对应高、低 ## "#FF0033","#1B9E77"
legend.title = i,#基因名写在图例题目的位置
font.legend = 11,#图例的字体大小
#太小的p value标为p < 0.001
pval = paste(pval = ifelse(p.val < 0.001, "p < 0.001",
paste("p = ",round(p.val,3), sep = "")),
HR, CI, sep = "\n"))
#每一个图保存为一个pdf文件
ggsave(paste0(i,".pdf"),width = 4,height = 4)


往期文章:
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
2. 精美图形绘制教程
- 精美图形绘制教程
3. 转录组分析教程
小杜的生信筆記,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!