欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/132047940
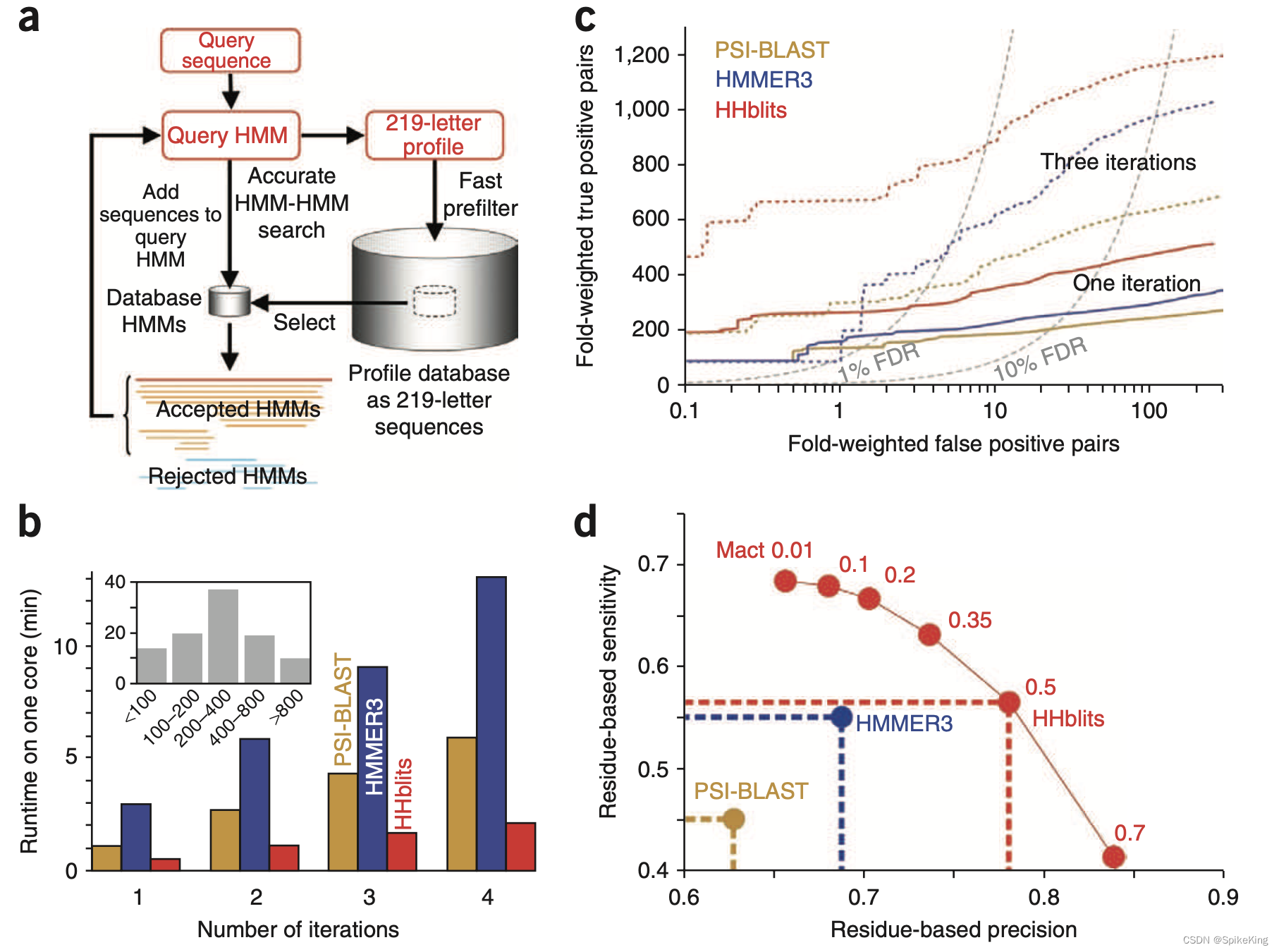
MMseqs2 与 HHblits 的算法比较:
- 蛋白质序列搜索算法 MMseqs2 与 HHblits 的搜索结果差异
- HHblits 算法搜索 BFD 与 UniRef30 的结果分析
Paper: HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment
- 2011.12 Nature Methods
HHblits 是一种基于 HMM-HMM 对齐的迭代蛋白质序列搜索工具,可以快速、灵敏、准确地找到数据库中与查询序列相似的蛋白质序列。HHblits 的主要特点有:
- 使用隐马尔可夫模型(HMM)来表示查询序列和数据库序列,而不是使用简单的氨基酸序列。HMM 是一种统计模型,可以捕捉序列的进化变化和保守区域,从而提高序列相似性的检测能力。
- 使用一种离散化的预过滤器,可以快速地筛选出与查询序列有潜在相似性的数据库序列,从而减少了 HMM-HMM 对齐的计算量。这种预过滤器可以将搜索时间提高 2500 倍,而不损失搜索灵敏度。
- 使用迭代的方法,可以利用上一轮搜索得到的多序列比对(MSA)来构建更精确的查询 HMM,来进行下一轮搜索。这样可以不断地扩展搜索范围,找到更多的相关序列。
在 bfd_uniref_hits.a3m 中,以 UniRef100 开头的序列描述,即来源于 UniRef30 数据库,即:
>UniRef100_A0A2X3SLP5 Uncharacterized protein n=2 Tax=Streptococcus equi TaxID=1336 RepID=A0A2X3SLP5_STRSZ
-TNtsSQEVDQVAQALELMFDNNVSTSNFKKYVNNNFSDSEIAIAELELESRISNSrsefrvawnemggCIAGKIRDEFFAMISVGTIVKYAQKKAWKELALVVLKFVKANGLKTNAIIVAGQLALWAVQCG
a3m文件格式是用于表示蛋白质序列比对的文本格式,是 FASTA 格式的一种扩展。在a3m文件格式中,蛋白质序列用单个字母表示,其中,匹配用大写字母表示,删除用横线(-) 表示,插入用小写字母表示,去掉小写字母,只保留大写字母与横线(-) ,则长度与目标序列相同。
其他序列就是来源于 BFD,以 SRR 开头的数量较多,即:
>SRR5690606_14355373
-----QIEELAAQLEFLMEEALiiENGERTfdfEKIENEFgkeVKDEIKMLTVDAQVWqvqpgaitlaanqPWKDCMVGAIKDHFGvALVTAaleGGLWAYLEKKAYKEAAKLLVKF----AVGTNAVGIAGTLIYYGGKCT
...
>SoimicmetaTmtLPB_FD_contig_61_1212631_length_209_multi_1_in_0_out_0_1 # 2 # 76 # 1 # ID=3042713_1;partial=10;start_type=Edge;rbs_motif=None;rbs_spacer=None;gc_cont=0.693
---NEEIEQLAADLEFLMEEAAiydEKGKVVnfdfDLLEERFgYVLELEMLKEEIEAYnattegd--NDeiqlfswksCMISALKGHFGvALIEValtGGLWSYLEKKAYKEAAKLLLKI----GIEGNVIGLTAFLTWYSVDCI
...
>APFre7841882654_1041346.scaffolds.fasta_scaffold441692_2 # 291 # 482 # 1 # ID=441692_2;partial=01;start_type=ATG;rbs_motif=AGGAG;rbs_spacer=5-10bp;gc_cont=0.609
-EIPLEAqgisiFGANHCDGareserliahelAHQWFgnsvtakrwrhiwlhegFACYAEWLWSEHSGdrsaDEWAhhfHEKLASSPQDLLLADPGPRDMFddrvykrgalTLHVLRRTLGDENFFALLKDWTSRHRHGSAVTD------DFTGLAANYTDQSLRPLWDAWLYS--TEVPALDAESX-------------------------------------------------------------------------------------------------------------------
...
因此,使用 MMseqs2 分别搜索 BFD 和 UniRef30,再合并,与使用 HHblits 一起搜索两个库的效果相同,同时也可以计算不同搜索算法的结果差异,即:
- 测试序列来源于 CASP15 提供的官方序列。
- MMseqs2 的搜索参数,
num_iterations是 3,sensitivity是 8,即i3s8,其余默认。 - HHblits 使用 AF2 默认的搜索工具,数据库也保持一致。
即:

源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2022. All rights reserved.
Created by C. L. Wang on 2023/8/1
"""
import os
from myutils.project_utils import read_file
from root_dir import DATA_DIR
class BfdUniref30Overlap(object):
"""
计算 MMseqs2 与 HHblits 搜索结果之间的重叠度
"""
def __init__(self):
pass
@staticmethod
def count_seqs(data_lines):
return len(data_lines) // 2 - 1
def split_hhblits(self, data_lines):
"""
拆分 Uniref30 与 BFD 数据
"""
n = len(data_lines)
n = n // 2 * 2
u_desc_list, u_seq_list = [], []
b_desc_list, b_seq_list = [], []
for i in range(2, n, 2):
desc = data_lines[i][1:]
seq = data_lines[i+1]
seq = seq.replace("-", "")
if desc.startswith("UniRef100"):
desc = desc.split(" ")[0]
u_desc_list.append(desc)
u_seq_list.append(seq)
else:
items = desc.split("|")
if len(items) > 1:
desc = items[1]
b_desc_list.append(desc)
b_seq_list.append(seq)
unique_u_seq = len(list(set(u_seq_list)))
unique_u_desc = len(list(set(u_desc_list)))
print(f"[Info] unique uniref30 desc: {unique_u_desc} / {len(u_desc_list)}")
print(f"[Info] unique uniref30 seqs: {unique_u_seq} / {len(u_seq_list)}")
unique_b_seq = len(list(set(b_seq_list)))
unique_b_desc = len(list(set(b_desc_list)))
print(f"[Info] unique bfd desc: {unique_b_desc} / {len(b_desc_list)}")
print(f"[Info] unique bfd seqs: {unique_b_seq} / {len(b_seq_list)}")
return u_desc_list, u_seq_list, b_desc_list, b_seq_list
def process(self, uniref30_path, bfd_path, hhblits_path):
assert os.path.isfile(uniref30_path) and os.path.isfile(bfd_path) and os.path.isfile(hhblits_path)
print(f"[Info] mmseqs uniref30: {uniref30_path}")
print(f"[Info] mmseqs bfd: {bfd_path}")
print(f"[Info] hhblits bfd and uniref30: {hhblits_path}")
uniref30_lines = read_file(uniref30_path)
bfd_lines = read_file(bfd_path)
hhblits_lines = read_file(hhblits_path)
print(f"[Info] uniref30: {self.count_seqs(uniref30_lines)}, bfd: {self.count_seqs(bfd_lines)}, "
f"hhblits: {self.count_seqs(hhblits_lines)}")
# ---------- 统计 MMseqs2 Uniref30 ---------- #
u_desc_list, u_seq_list = [], []
n = len(uniref30_lines)
n = n // 2 * 2
for i in range(2, n, 2):
u_name = uniref30_lines[i][1:].split("\t")[0]
u_seq = uniref30_lines[i+1].replace("-", "")
assert u_name.startswith("UniRef100")
u_desc_list.append(u_name)
u_seq_list.append(u_seq)
assert len(u_desc_list) == self.count_seqs(uniref30_lines)
unique_u_seq = len(list(set(u_seq_list)))
unique_u_desc = len(list(set(u_desc_list)))
print(f"[Info] unique uniref30 desc: {unique_u_desc} / {len(u_desc_list)}")
print(f"[Info] unique uniref30 seqs: {unique_u_seq} / {len(u_seq_list)}")
# ---------- 统计 MMseqs2 Uniref30 ---------- #
# ---------- 统计 BFD Uniref30 ---------- #
b_desc_list, b_seq_list = [], []
n = len(bfd_lines)
n = n // 2 * 2
for i in range(2, n, 2):
b_name = bfd_lines[i][1:].split("\t")[0]
b_seq = bfd_lines[i+1].replace("-", "")
b_desc_list.append(b_name)
b_seq_list.append(b_seq)
assert len(b_desc_list) == self.count_seqs(bfd_lines)
unique_b_seq = len(list(set(b_seq_list)))
unique_b_desc = len(list(set(b_desc_list)))
print(f"[Info] unique bfd desc: {unique_b_desc} / {len(b_desc_list)}")
print(f"[Info] unique bfd seqs: {unique_b_seq} / {len(b_seq_list)}")
# ---------- 统计 BFD Uniref30 ---------- #
# ---------- 统计 HHblits BFD and Uniref30 ---------- #
hu_desc_list, hu_seq_list, hb_desc_list, hb_seq_list = self.split_hhblits(hhblits_lines)
assert len(hu_desc_list) + len(hb_desc_list) == self.count_seqs(hhblits_lines)
# ---------- 统计 HHblits BFD and Uniref30 ---------- #
# ---------- 统计 交集 ---------- #
num_u_desc_is = len(set(u_desc_list).intersection(set(hu_desc_list)))
print(f"[Info] uniref30 desc intersection: {num_u_desc_is}")
num_u_seqs_is = len(set(u_seq_list).intersection(set(hu_seq_list)))
print(f"[Info] uniref30 seqs intersection: {num_u_seqs_is}")
num_b_desc_is = len(set(b_desc_list).intersection(set(hb_desc_list)))
print(f"[Info] bfd desc intersection: {num_b_desc_is}")
num_b_seqs_is = len(set(b_seq_list).intersection(set(hb_seq_list)))
print(f"[Info] bfd seqs intersection: {num_b_seqs_is}")
# ---------- 统计 交集 ---------- #
# ---------- 统计 总数 ---------- #
unique_mmseqs_seqs = list(set(u_seq_list + b_seq_list))
unique_hhblits_seqs = list(set(hu_seq_list + hb_seq_list))
print(f"[Info] unique_mmseqs_ses: {len(unique_mmseqs_seqs)}")
print(f"[Info] unique_hhblis_ses: {len(unique_hhblits_seqs)}")
# ---------- 统计 总数 ---------- #
def main():
fasta_name = "T1104-D1_A117"
# fasta_name = "T1137s8-D1_A251"
# fasta_name = "T1188-D1_A573"
# fasta_name = "T1157s1_A1029"
uniref30_path = os.path.join(DATA_DIR, "overlap", fasta_name, f"{fasta_name}-uniref30-i3s8.a3m")
bfd_path = os.path.join(DATA_DIR, "overlap", fasta_name, f"{fasta_name}-bfd-i3s8.a3m")
hhblits_path = os.path.join(DATA_DIR, "overlap", fasta_name, "bfd_uniref_hits.a3m")
buo = BfdUniref30Overlap()
buo.process(uniref30_path, bfd_path, hhblits_path)
if __name__ == "__main__":
main()


![P1219 [USACO1.5] 八皇后 Checker Challenge](https://img-blog.csdnimg.cn/3b425915f3474cce816069b5b5a74f1f.png#pic_center)