学习目标
-
了解模型拟合的过程 -
比较两种假设检验方法(Wald test vs. LRT) -
了解多重测试校正的重要性 -
了解用于多重测试校正的不同方法
1. 模型拟合和假设检验
DESeq2 工作流程的最后一步是对每个基因进行计数并将其拟合到模型中并测试差异表达。
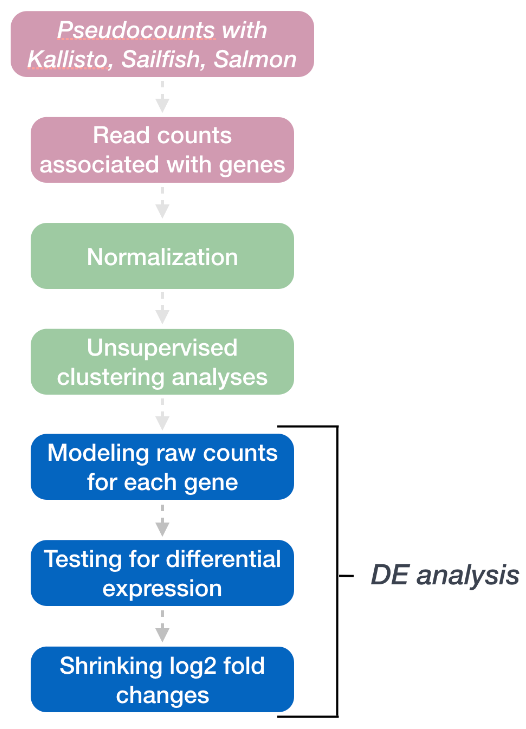
2. 广义线性模型
如前所述,RNA-seq 生成的计数数据表现出过度分散(方差 > 均值),用于对计数建模的统计分布需要考虑到这一点。因此,DESeq2 使用负二项分布通过以下公式对 RNA-seq 计数进行建模:
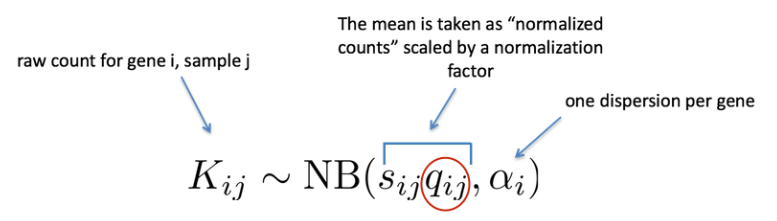
所需的两个参数是size factor和dispersion estimate。接下来使用广义线性模型 (GLM) 来拟合数据。建模是一种数学形式化的方法,用于在给定一组参数的情况下估算数据的方式。模型拟合后,将估计每个样本组的系数及其标准误差。系数是 log2 倍数变化的估计值,将用作假设检验的输入。
3. 假设检验
假设检验的第一步是为每个基因建立一个零假设。在我们的例子中,原假设是两个样本组之间没有差异表达 (LFC == 0)。然后,我们使用统计检验来根据观察到的数据确定原假设是否为真。
3.1. Wald test
在 DESeq2 中,Wald 检验是比较两组时用于假设检验的默认值。 Wald 检验是通常对已通过最大似然估计的参数执行的检验。在我们的案例中,我们正在测试每个基因模型系数 (LFC),这些系数是使用分散等参数得出的,这些参数是使用最大似然估计的。
DESeq2 通过以下方式实施 Wald 测试:
-
取 LFC 并将其除以标准误差,得到 z 统计量 -
将 z 统计量与标准正态分布进行比较,并计算 p 值,报告随机选择至少与观察值一样极端的 z 统计量的概率 -
如果 p 值很小,我们拒绝零假设并声明有证据反对零假设(即基因差异表达)
模型拟合和 Wald 检验先前已作为 DESeq() 函数的一部分运行:
# 以下仅作示例,上一个教程已经运行
dds <- DESeqDataSetFromTximport(txi, colData = meta, design = ~ sampletype)
dds <- DESeq(dds)
3.2. 似然比检验
当比较两个以上的样本类别时,DESeq2 还提供似然比检验 (LRT) 替代假设检验。LRT 不是评估一个基因的表达在一个类别中相对于另一个类别是上调还是下调,而是识别在不同样本类别中在任何方向上表达发生变化的基因。
-
这与 Wald 检验相比如何?
Wald 检验(默认)仅估计每个基因一个模型并评估 LFC == 0 的原假设。
对于似然比检验,还对已通过最大似然估计的参数执行。对于这个测试,每个基因估计两个模型;将一个模型的拟合度与另一个模型的拟合度进行比较。
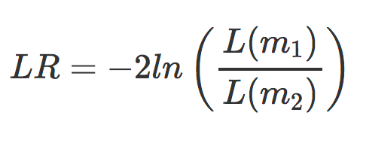
-
m1 是简化模型(即删除主要因素项的设计公式) -
m2 是完整模型(即您在创建 dds对象时提供的完整设计公式)
在这里,我们正在评估完整模型与简化模型一样适合的原假设。如果我们拒绝零假设,这表明完整模型(以及我们感兴趣的主要因素)解释了大量变异,因此该基因在不同水平上差异表达。 DESeq2 通过使用偏差分析 (ANODEV) 来比较两个模型拟合来实现 LRT。结果表明,LR 服从卡方分布,这可用于计算和关联的 p 值。
要使用 LRT,我们使用 DESeq() 函数,但这次添加两个参数:
-
指定我们要使用 LRT 测试 -
“简化”模型
# Likelihood ratio test
dds_lrt <- DESeq(dds, test="LRT", reduced = ~ 1)
由于我们的“完整”模型只有一个因素(样本类型),“简化”模型(删除该因素)在我们的设计公式中没有留下任何东西。 DESeq2 无法拟合设计公式中没有任何内容的模型,因此在没有其他协变量的情况下,截距使用语法 ~ 1 建模。
4. Multiple test correction
无论我们使用 Wald 检验还是 LRT,每个经过检验的基因都会与一个 p 值相关联。我们正是用这个结果来确定哪些基因被认为是显著差异表达的。但是,我们不能直接使用 p 值。
4.1. p-value
显著性截断值 p < 0.05 的基因意味着它有 5% 的几率是假阳性。例如,如果我们测试 20,000 个基因的差异表达,在 p < 0.05 时,我们预计会偶然发现 1,000 个基因。如果我们发现总共有 3000 个基因存在差异表达,那么大约三分之一的基因是假阳性!
由于每个 p 值都是单个测试(单个基因)的结果。我们测试的基因越多,我们的假阳性率就越大。这就是多重检验问题。
4.2. 校正
多重检验有几种常见的校正方法:
-
Bonferroni:调整后的 p 值计算方式为:p 值 * m(m = 测试总数)。这是一种非常保守的方法,假阴性的可能性很高,因此通常不推荐使用。 -
FDR/Benjamini-Hochberg: Benjamini 和 Hochberg (1995) 定义了错误发现率 (FDR) 的概念,并创建了一种算法,以在给定独立 p 值列表的情况下将预期 FDR 控制在指定水平以下。 -
Q-value / Storey method: 称该特征为重要时可达到的最低 FDR。例如,如果基因 X 的 q 值为 0.013,则表示 p 值至少与基因 X 一样小的基因中有 1.3% 是假阳性。
DESeq2 通过去除那些在测试前不太可能显著 DE 的基因,例如那些具有低计数和异常样本(基因级 QC)的基因,帮助减少测试的基因数量。但是,还实施了多重测试校正,以使用 Benjamini-Hochberg 程序的解释来降低错误发现率。
4.3. FDR < 0.05
通过将 FDR 截止值设置为 < 0.05,我们是说我们预期差异表达基因中的假阳性比例为 5%。例如,如果您将 500 个基因称为差异表达,FDR 截断值为 0.05,您预计其中 25 个是假阳性。
欢迎Star -> 学习目录
更多教程 -> 学习目录
本文由 mdnice 多平台发布