生成虚拟数据集
library(ggplot2)
data.matrix <- matrix(nrow = 100, ncol = 10)
colnames(data.matrix) <- c(
paste("wt",1:5,sep = ""),
paste("ko",1:5,sep = "")
)
rownames(data.matrix) <- paste("gene",1:100,sep = "")
head(data.matrix)
以上代码生成了100行基因,10列样本的矩阵
前五列命名wt开头+1-5,表示正常基因
后五列命名ko开头+1-5,表示缺少基因的样本(knock-out)
给每行基因都统一命名gene+1-100
head()函数默认查看前6行

现在只是定义了矩阵的shape和name,还没填充数值
for (i in 1:100){
wt.values <- rpois(5, lambda = sample(x=10:1000, size = 1))
ko.values <- rpois(5, lambda = sample(x=10:1000, size = 1))
data.matrix[i,] <- c(wt.values, ko.values)
}
head(data.matrix)
这段代码的作用是生成一个大小为100x10的数据矩阵data.matrix,其中前5列是"wt"(wild-type)样本的值,后5列是"ko"(knockout)样本的值。
在循环中,对于每个i的取值(从1到100),首先使用sample(x=10:1000, size = 1)从10到1000之间的整数中随机抽取一个数作为泊松分布的参数lambda。然后,使用rpois(5, lambda)函数生成一个具有泊松分布的随机数向量,其中每个元素表示一个基因在"wt"样本中的表达量。同样的过程也用于生成"ko"样本中的表达量。
最后,通过c(wt.values, ko.values)将"wt"和"ko"样本的表达量合并为一个长度为10的向量,并将其赋值给data.matrix的第i行。
用for循环给依次给1-100行的前五列和后五列赋值,填充值介于10-1000之间。

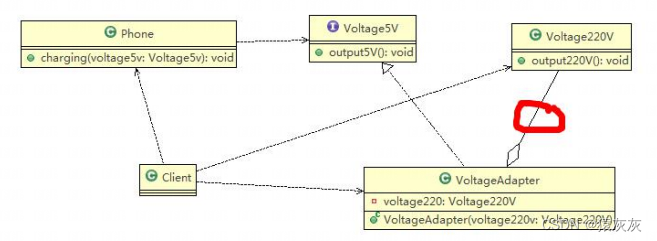
初始虚拟数据集创建完毕,接下来用prcomp()函数分析各样本之间关系。该函数默认情况下以基因为列,样本为行,和我们创建的矩阵互为转置,因此需要用到转置函数t()
pca <- prcomp(t(data.matrix), scale = TRUE)
plot(pca$x[,1], pca$x[,2])
pca.var <- pca$sdev^2
pca.var.per <- round(pca.var/sum(pca.var)*100, 1)
barplot(pca.var.per, main = "Screen Plot", xlab ="principal component", ylab = "percent variation")
prcomp(, scale = TRUE)表示对数据进行标准化
每一列表示一个基因所对应的10个样本,即一列只有十个数据
plot 生成一个2D的图,前两个主成分的散点图

pca.var 表示 标准差的平方
pca.var.per 表示 每个变量所占的百分比,保留小数点后一位

可以看到前两个成分所占比例最大,尤其是第一个成分
用 barplot 来直观每个成分所占比例

ggplot2绘图
pca.data <- data.frame(sample = rownames(pca$x),
X = pca$x[,1],
Y = pca$x[,2]
)
ggplot(data = pca.data, aes(x = X, y = Y,label = sample))+
geom_text()+
xlab(paste("pc1-", pca.var.per[1], "%", sep = ""))+
ylab(paste("pc2-", pca.var.per[2], "%", sep = ""))+
theme_bw()+
ggtitle("my pac graph")
先按 ggplot2 需要的方式格式化数据,x轴用第一个成分,y轴用第二个成分

可以发现数据分布在了两侧,我们用prcomp()调用负载得分loading scores 的参数rotation
通过分析pca$rotation,可以了解该主成分与哪些基因相关性较高,哪些基因对主成分的贡献较大。这对于解释主成分分析的结果和理解数据的结构和变化模式非常有帮助。
loading_scores <- pca$rotation[,1]
gene_score <- abs(loading_scores)
gene_score_ranked <- sort(gene_score,decreasing = TRUE)
top_10_genes <- names(gene_score_ranked[1:10])
pca$rotation[top_10_genes, 1]
这里我们查看 PC1 的负载得分,因为 PC1 解释原始数据的 93.6%的方差
loading_scores <- pca$rotation[,1] 这行代码的作用是将PCA分析结果中第一个主成分(即第一列)的负载得分(即100个基因数据)提取出来并赋值给loading_scores变量。

abs()取绝对值,再从小大到小排序,选取排名靠前的前十个基因top_10_genes

若想显示每个基因对应的 rotation,用以下代码即可,展示的是带正负值的
pca$rotation[top_10_genes, 1]
排名前10的基因在第一主成分上的负载得分
总的来说,本文对基因表达数据进行了主成分分析,并可视化结果。
通过PCA主成分分析,可以降维并找到影响数据变化最大的主要因素,进而进行数据的可视化和分析。




![[安洵杯 2019]game](https://img-blog.csdnimg.cn/1695ff85417445fea7616eefd958eea1.png)