****Monocle2全部往期精彩系列:
1、群成员专享:Monocle2更新(就是重新梳理一下)
2、一键跑完monocle2?
3、ggplot2个性可视化monocle2结果
4、ggplot修饰monocle2拟时热图:一众问题全部解决
5、Monocle2终极修改版
6、单细胞拟时分析:基因及通路随拟时表达变化趋势
Monocle2拟时分析及可视化我们整了好多呀,算是整明白了吧。上一期我们做了Monocle3的拟时基因富集分析(Monocle3个性化分析作图:拟时热图/拟时基因GO分析/拟时基因趋势分析)。Monocle2的还没有做过,这里我们演示下,其实方法是一样的。monocle2拟时热图修饰我们已经做过了。这里的演示我们不再使用修饰,用monocle2本来的拟时热图绘制函数进行,然后通过聚类不同的module提取基因进行富集分析。本贴涉及到几个修改函数和自写函数的加载,数据代码已上传QQ群! 接下来我们就具体看看做法,首先还是做热图。这里我们用R包里面的plot_pseudotime_heatmap函数即可。
#以下是monocle2做热图的代码,很常规的操作
library(monocle)
library(Seurat)
library(dplyr)
library(viridis)
library(pheatmap)
library(grid)
#======================================================================================
setwd("D:/KS项目/公众号文章/monocle2拟时热图富集分析")
mouse_monocle <- readRDS("D:/KS项目/公众号文章/monocle2拟时结果个性化作图/mouse_monocle.rds")
peu_gene <- differentialGeneTest(mouse_monocle,fullModelFormulaStr = "~sm.ns(Pseudotime)",cores = 2)
write.csv(peu_gene,file='peu_gene.csv')#保存好文件,这个分析过程挺费时间
peu_gene <- read.csv('peu_gene.csv', header = T, row.names = 1)
peu_gene <- peu_gene[which(peu_gene$qval<0.01 & peu_gene$num_cells_expressed>100),]#筛选显著是的
peu_gene %>% arrange(qval) -> peu_gene#按照qval排个序
peu_gene <- peu_gene[1:100,] #这里我们取前100个基因演示
#热图的做法就很多了,可以用它默认的函数,也可以用我们讲过的ggplot或者pheatmap自己修饰
p <-plot_pseudotime_heatmap(mouse_monocle[peu_gene$gene_short_name,],
num_clusters = 4,
cores = 2,
show_rownames = T,return_heatmap =T,
hmcols = viridis(256),
use_gene_short_name = T)
p
class(p)
# [1] "pheatmap"

image.png
plot_pseudotime_heatmap函数好像并不能选择性展示感兴趣的基因,然而提取数据用pheatmap或者ggplot作图我们也做过了,这里再去修饰一遍展示没有必要,我就想用monocle2包热图函数完成。class一下plot_pseudotime_heatmap函数的产生的图,发现是pheatmap对象,查看作者的原函数也发现这个热图作者是基于pheatmap做的,那就好办了,如果连续阅读我们帖子的小伙伴可能会想到,我们之前写过一篇帖子:学习nature medicine函数:热图标记任意基因,这里面我们引用了一个文章提供的函数,可以很方便的为pheatmap标记基因。。然而呢,由于上图是分裂的,所以这个函数效果不好,那么办法要么就是修改这个添加基因的函数,另一个方法就是修改plot_pseudotime_heatmap函数。权衡之下,偷懒的我选择了修改plot_pseudotime_heatmap函数,因为它好修改!
#改造热图函数,展示需要的基因
source('./new_heatmap.R')
p1 <-plot_pseudotime_heatmap(mouse_monocle[peu_gene$gene_short_name,],
num_clusters = 4,
cores = 2,
show_rownames = T,return_heatmap =T,
hmcols = viridis(256),
use_gene_short_name = T)
p1

image.png
这样就既有基因分组注释,但是热图行不会分裂开。下面使用add.flag.R函数标记感兴趣基因。
#只展示感兴趣的基因
genes <- c("Cxcr2","Celf2","Gsr","Sorl1","Ftl1","Ptprc",
"Cst3","Il1b","Fgl2","Sirpb1c","Maml3","Peak1",
"Orm1","H2afz","Cpa6","Ica1","Fmnl2","Cpne3",
"Anxa3","Tbc1d8","Camp","Mmp8","S100a8","S100a9")
source('./add.flag.R')
add.flag(p,kept.labels = genes,repel.degree = 0.2)

image.png
接下来就是提取module基因进行富集分析了,我们直接包装为一个函数Monocle2_gene_enrichment.R,直接运行即可,当然有个限制我们只写了GO富集分析,感兴趣可以自己添加KEGG或者其他的分析。
#提取module基因,进行富集分析
library(clusterProfiler)
library(ggplot2)
source('./Monocle2_gene_enrichment.R')
GOannlysis <- Monocle2_gene_enrichment(p,knum=4,species='org.Mm.eg.db',
pvalueCutoff=0.05,
qvalueCutoff=0.05)
table(GOannlysis$Cluster)#module1基因最少,没有显著性富集
# 1 2 3 4
# 0 76 43 77
write.csv(GOannlysis, file = 'GOannlysis.csv')
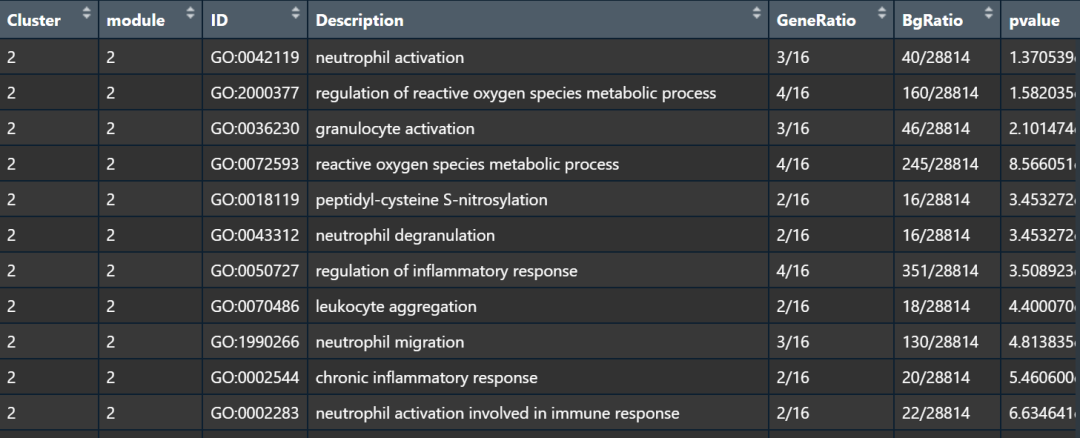
image.png
最后,将热图在AI中修饰,旁边展示富集通路,这个图就完整了。好了,这就是monocle2的全部内容了,觉得分享对您有帮助的,点个赞分享下再走呗!