Gaussian是做半经验计算和从头计算使用最广泛的量子化学软件,可研究诸如分子轨道,结构优化,过渡态搜索,热力学性质,偶极矩和多极矩,电子密度和电势,极化率和超极化率,红外和拉曼光谱,NMR,垂直电离能和电子亲合能,化学反应机理,势能曲面和激发能 QM/MM计算等化学领域的许多课题。应用非常广泛,而且易于上手。
LAMMPS是一款经典的分子动力学软件,免费开源,可以模拟液态、固态或气态的粒子的系综。也可以采用不同的力场和边界条件来模拟全原子,聚合物,生物,固态(金属、陶瓷,氧化物),粒状和粗料化体系。LAMMPS可以计算的体系小至几个粒子,大到上百万甚至是上亿个粒子。同时lammps代码可以修改和扩展,可以方便的为之扩展上新特征和功能来匹配课题的个性化需求。
Gaussian量子化学计算技术与应用
2023年06月23日-06月24日
2023年07月01日-07月02日
①.基础入门
②.案例实操进阶(结构几何优化,单点能(能量)分析)(分子轨道、轨道能级计算)(势能面相关计算)(各类光谱计算及绘制)(激发态专题、高精度和多尺度计算方法)Gaussian报错及其解决方案等
专题二:lammps及Reaxff反应力场二合一专题
2023年06月22日-06月24日
2023年07月01日-07月02日
①.基础入门
②.案例实操进阶(石墨烯,金属材料模拟及力学分析)(纳米流体模拟及流动性质分析)(热传导及导热系数模拟计算)(多成分体系模拟)(离子辐照对石墨烯、金属、碳化硅的离位损伤模拟)
③自建分子力场参数文件和金属有机框架材料晶体模型(创建及MOFs材料建模)
④分子筛纳米膜分离H2/CO2混合气体模拟
⑤利用ReaxFF模块研究碳氢化合物的燃烧
⑥利用ReaxFF模块研究化学机械抛光

Gaussian量子化学计算、LAMMPS分子动力学模拟
news2026/2/13 14:46:36
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.coloradmin.cn/o/559410.html
如若内容造成侵权/违法违规/事实不符,请联系多彩编程网进行投诉反馈,一经查实,立即删除!相关文章

超越竞争的获客之道:DTC品牌出海策略全面解析
随着全球数字化的快速发展,DTC品牌正迎来一个全新的时代。然而,随着越来越多的DTC品牌进入国际市场,如何在激烈的竞争中脱颖而出,并获得新客户成为一个关键的挑战。本文Nox聚星将和大家深入探讨DTC品牌在出海时代如何破解获客困局…
外包干了五年,废了...
先说一下自己的情况。大专生,17年通过校招进入湖南某软件公司,干了接近5年的测试点点点,今年年上旬,感觉自己不能够在这样下去了,长时间呆在一个舒适的环境会让一个人堕落!而我已经在一个企业干了五年的点工…
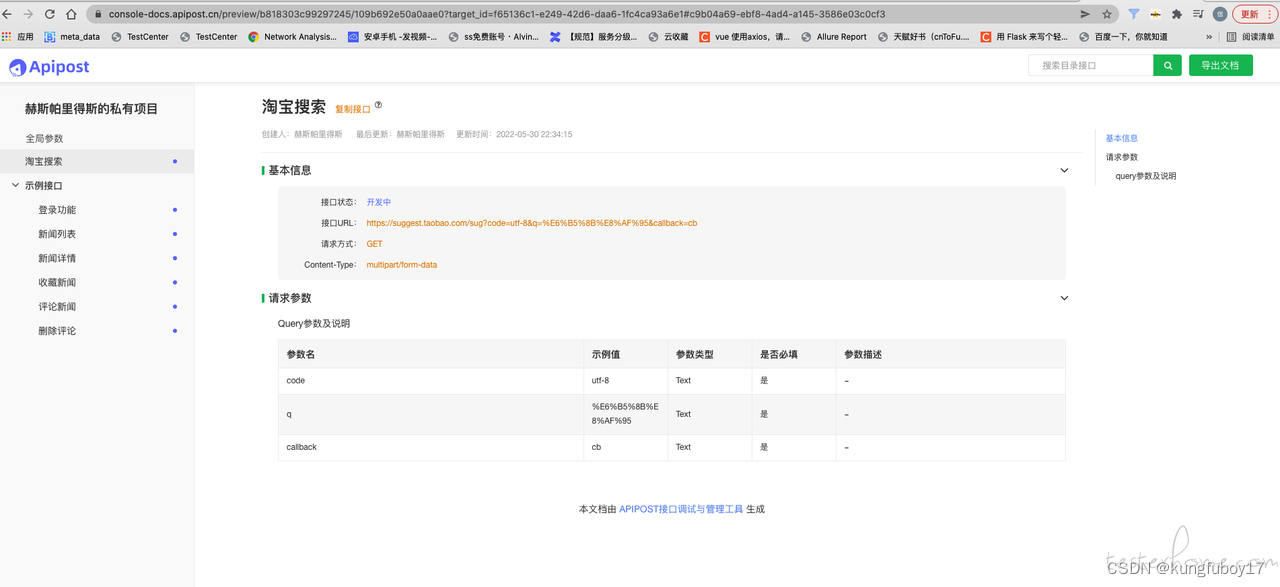
10年开发,浅谈eolink 、aifox、apipost 横向对比
功能对比
在实际工作中,eolink 、apifox、apipost 三个工具,我个人都有使用。接下来,我会对三款功能对比,于是我拉了个功能对比的清单。 特别说明:以下的对比,不吹不黑,只列功能,纯客观比对,不带有任何商业带货的意义。 eolink 、aifox、apipost 三款工具有很多功能模块,本次仅…
acwing提高——DFS之连通性问题+搜索顺序
1 连通性问题(内部搜索)
内部搜索一般不用恢复现场
1.迷宫
题目http://ybt.ssoier.cn:8088/problem_show.php?pid1215
#include<bits/stdc.h>
using namespace std;
const int N110;
bool st[N][N];
char g[N][N];
int n;
int sx,sy,ex,ey;
…

95后字节八年测开晒出工资单:狠补了这个,真香···
最近一哥们跟我聊天装逼,说他最近从字节跳槽了,我问他跳出来拿了多少?哥们表示很得意,说跳槽到新公司一个月后发了工资,月入5万多,表示很满足!这样的高薪资着实让人羡慕,我猜这是税后…

2022年深圳杯数学建模B题基于用电可靠性的配电网规划解题全过程文档及程序
2022年深圳杯数学建模
B题 基于用电可靠性的配电网规划
原题再现: 如果一批用户变压器(下面简称用户)仅由一个电源变电站(下面简称电源)供电,称为单供。这时配电网由电线和开关联接成以电源为根节点的树状…
k8s使用ECK形式部署elasticsearch+kibana
文章目录 前言一、ECK是什么?二、安装ECK1.crd.yaml2.operator.yaml 三、安装es,elasticsearch-cluster.yaml四、安装kibana总结 前言
在k8s上基于ECK(2.4)部署elasticsearch,简单记录一下,主要是quicksta…

《程序员面试金典(第6版)》面试题 02.03. 删除中间节点(特殊的删除节点操作)
题目描述 若链表中的某个节点,既不是链表头节点,也不是链表尾节点,则称其为该链表的「中间节点」。 题目传送门:面试题 02.03. 删除中间节点 假定已知链表的某一个中间节点,请实现一种算法,将该节点从链表中…
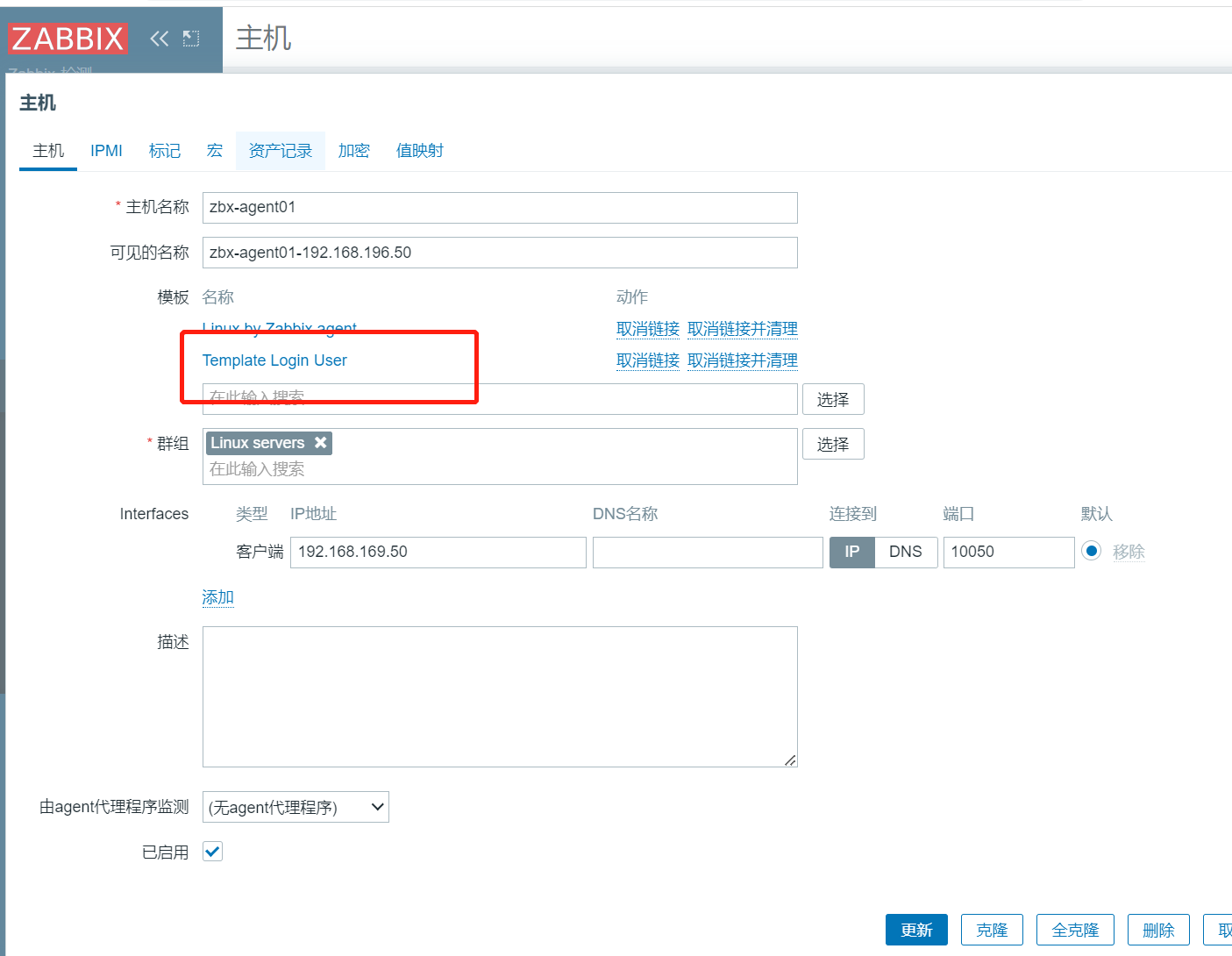
服务(第二十九篇)zabbix
zabbix 是什么?
zabbix 是一个基于 Web 界面的提供分布式系统监视以及网络监视功能的企业级的开源解决方案。 zabbix 能监视各种网络参数,保证服务器系统的安全运营;并提供灵活的通知机制以让系统管理员快速定位/解决存在的各种问题。 zabbi…

太阳升起和落下(长文警告)
今天分享一个太阳升起落下的动画场景。
有朋友问我为什么只发代码不做说明,今天我们尝试下对代码进行注解说明一下。 首先这个场景大致的可拆分为4个部分:太阳/月亮,右下角的按钮,天上的云和最显眼的建筑。 我们先做一个按钮&am…
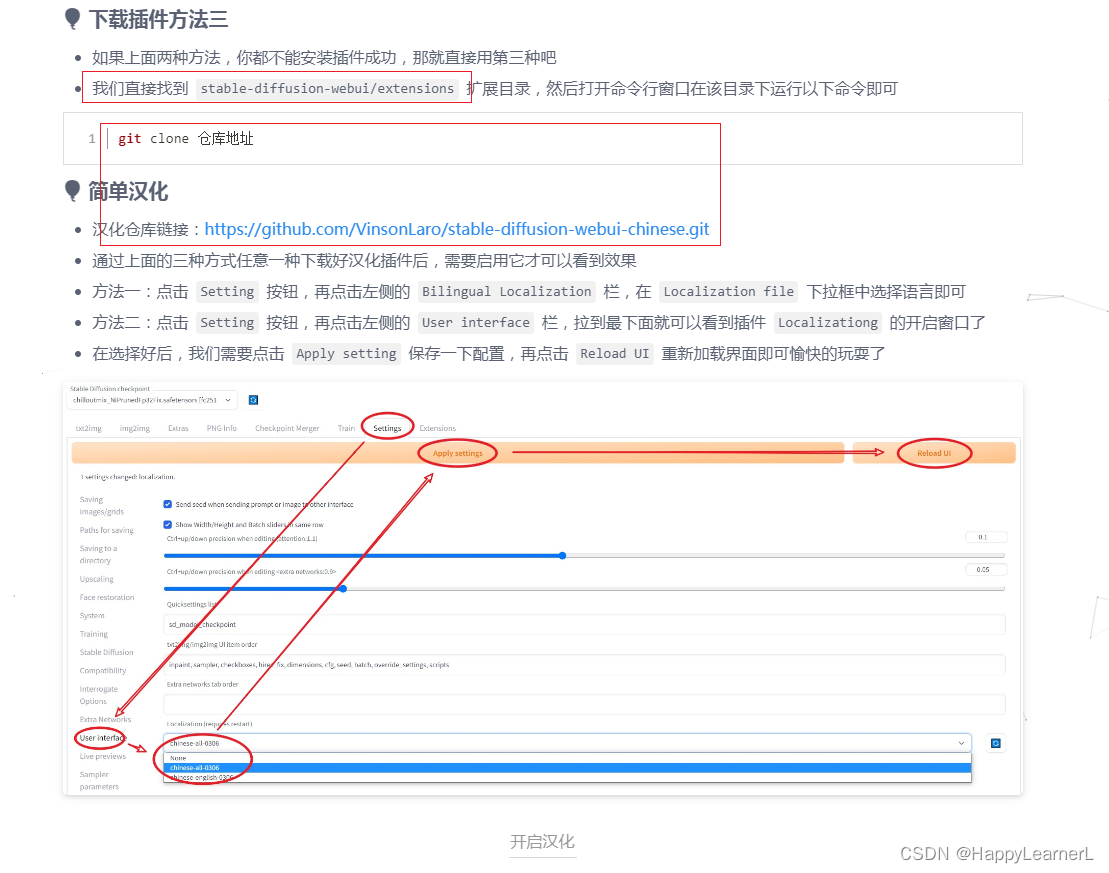
stable diffusion AI绘图工具的安装和使用centos7.8系统
stable diffusion 作图工具本地部署 重要【AI作画】stable diffusion webui Linux虚拟机 Centos 详细部署教程 服务器CentOS 7 安装 Stable Diffusion WebUI ,并映射到本地浏览器 CentOs7 Stable Diffusion Novel AI实现AI绘画 stable diffusion webui安装部署…
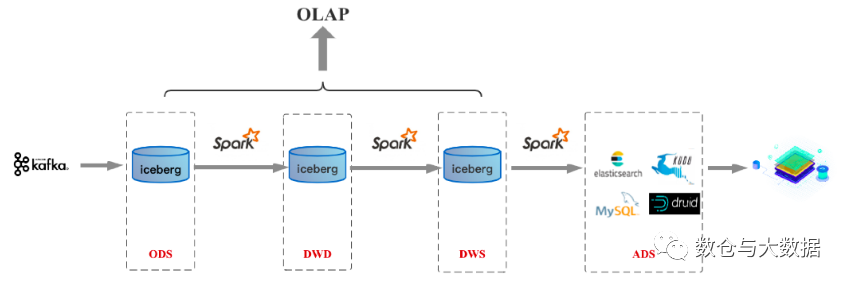
二、数据仓库详细介绍
基础概念 架构与框架,架构是结构,框架是规范 模块与组件,模块是逻辑概念,通过分解使复杂问题简单化,组件是物理概念,将具体的模块落地,且各个组件间保持松散耦合 定义:架构&#x…

分布式全局唯一id实现总结
前言:本文意在对借助db和程序生成分布式id进行一些总结,以及对其特性进行比较分析;
1 实现方式:
Db 通过配置步长和初始值的方式,使得每个db库生成id 的不同性,如 3个db 实例情况下:其步长均设…

我3年前写的博客,又被别人抄去发论文了,该论文整个正文部分几乎直接照抄我的博客
我想说每一篇原创博客都是作者的心血,有时候写一篇博客也许会花一天,甚至好几天的时间,尊重原创,营造好的环境,才有可能出现更多优质的博文,而不是到处都是抄来抄去的低质量水文。 前几天接到来自粉丝的私信…
加密解密软件VMProtect教程(八)许可制度之集成到应用程序
VMProtect是新一代软件保护实用程序。VMProtect支持德尔菲、Borland C Builder、Visual C/C、Visual Basic(本机)、Virtual Pascal和XCode编译器。
同时,VMProtect有一个内置的反汇编程序,可以与Windows和Mac OS X可执行文件一起…
全网最快PCB打样| 急速小批量打样的秘密在这里
对于广大爱好者以及电子工程师来说,电子行业的快速发展,导致电子产品的多样化和个性化,电路设计的复杂度和难度提高,需要更多的PCB打样来验证和测试,满足细分市场的客户需求。 随着PCB打样厂家的服务优化和价格降低&am…
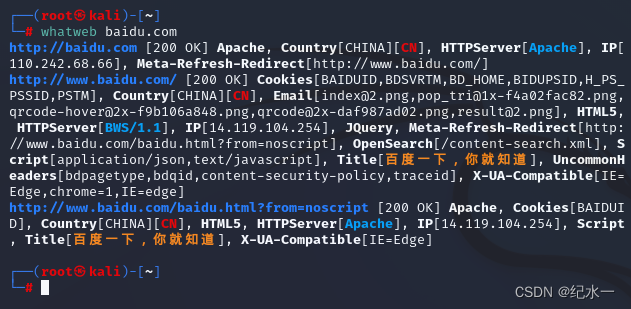
信息收集-服务器信息
服务器上面可以运行大量的系统服务和第三方应用服务,如果操作系统或者第三方软件没有及时升级打补丁,攻击者就有可能直接通过服务器上运行的服务进行攻击。
服务器需要收集的信息包含三个方面:
操作系统信息等识别waf(Web应用程…
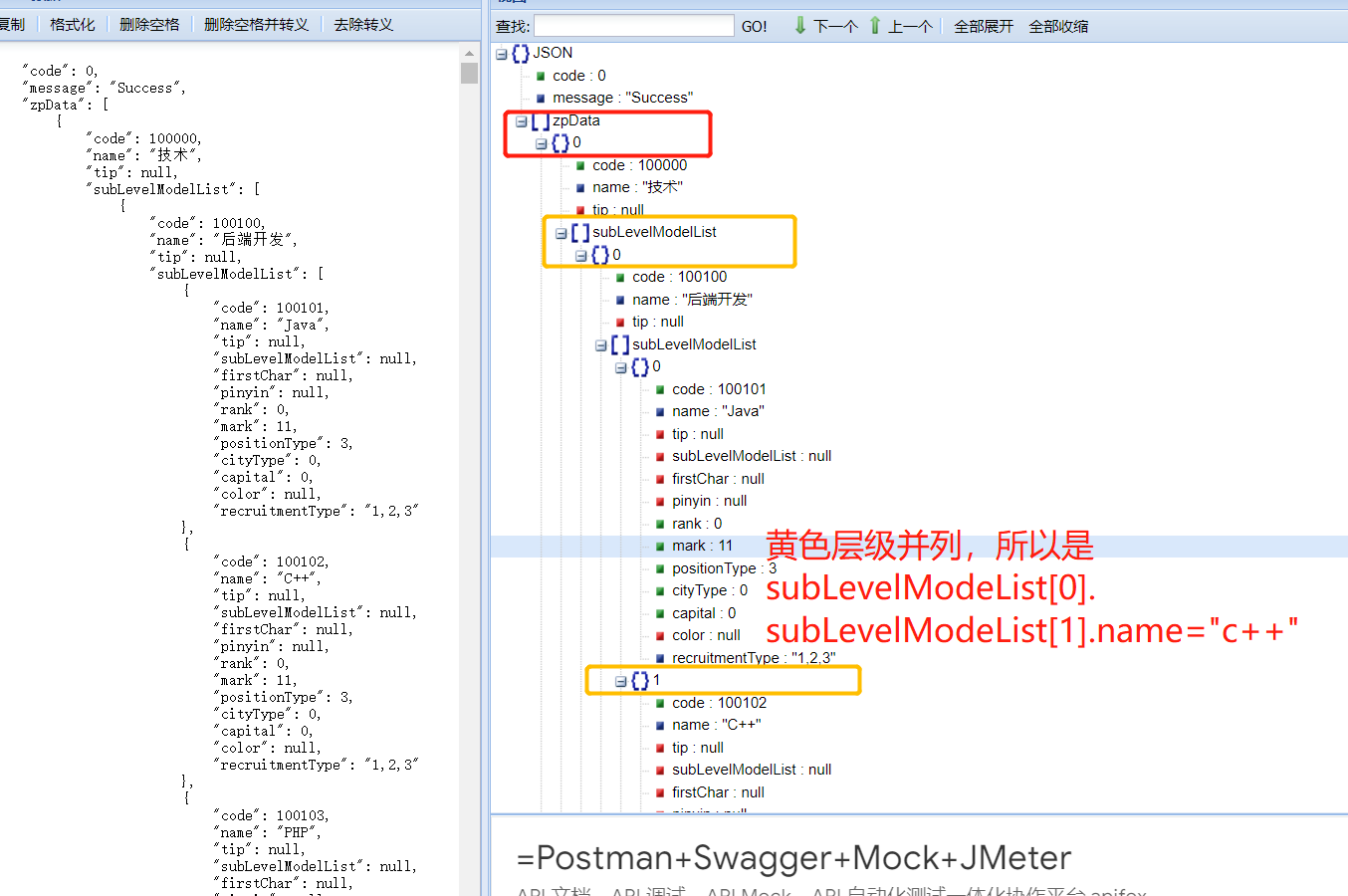
高级测试必备技能:从session请求到token请求,傻瓜式掌握Charles和postman断言技巧
引言
在快速发展的数字化时代,软件测试作为保障软件品质和用户体验的重要一环,显得愈加重要。SESSION请求、TOKEN请求、charles使用和Postman断言等技术,成为测试人员不可或缺的必备技能。
在这篇文章中,我将深入浅出地为您讲解…

Java编程思想(第4版) 扫描版
Java编程思想 - 基础必备
Java基础必备书籍!
从本书获得的各项大奖以及来自世界各地的读者评论中,不难看出这是一本经典之作。本书的作者拥有多年教学经验,对C、C以及Java语言都有独到、深入的见解,以通俗易懂及小而直接的示例解…
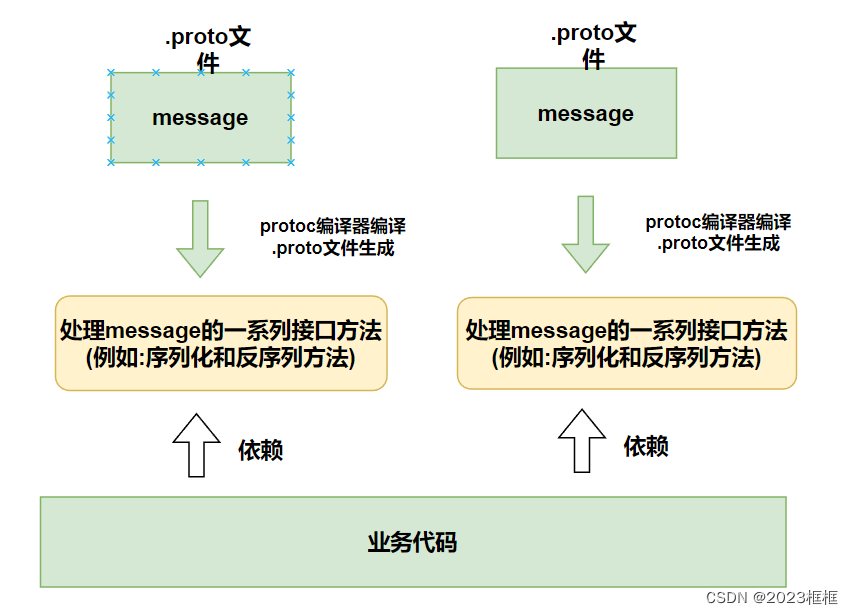
使用C++快速上手ProtoBuf (一)
文章目录 课程目标一、初始ProtoBuf1. 序列化概念2.ProtoBuf是什么3.ProtoBuf的使⽤特点 二、安装ProtoBuf三、教学思路四、快速上⼿步骤1:创建.proto文件步骤2:编译contacts.proto⽂件,⽣成C⽂件步骤3:序列化与反序列化的使⽤⼩结…