谷禾健康

微生物组的纵向研究是一种长期跟踪微生物组变化的研究方法。在这类研究中,样本从同一人群或个人中多次采集,通过检测样本中微生物群落丰度的变化(如不同菌群的比例和种类),来了解微生物组随时间的变化趋势和特征。
根据微生物组不同菌群的变化,可以发现一些与健康或疾病相关的特征,从而为疾病预防和治疗提供参考。
但是,由于不同研究对象在纵向时间轴上的时间点分布不均匀且数量不同,难以进行有效的综合分析,一些时间点上的样本缺失导致大量数据无法利用。
为此,研究人员提出了一种名为DeepMicroGen的双向循环神经网络-生成对抗网络(GAN)模型,该模型根据观测值之间的时间关系进行训练,从而在时间序列数据中填补缺失值。
本文介绍了DeepMicroGen模型的算法、并在性能评估上使用模拟和真实数据集与几种标准基线方法进行了比较,结果都表示DeepMicroGen模型无疑是优秀的。
DeepMicroGen模型训练方法概述

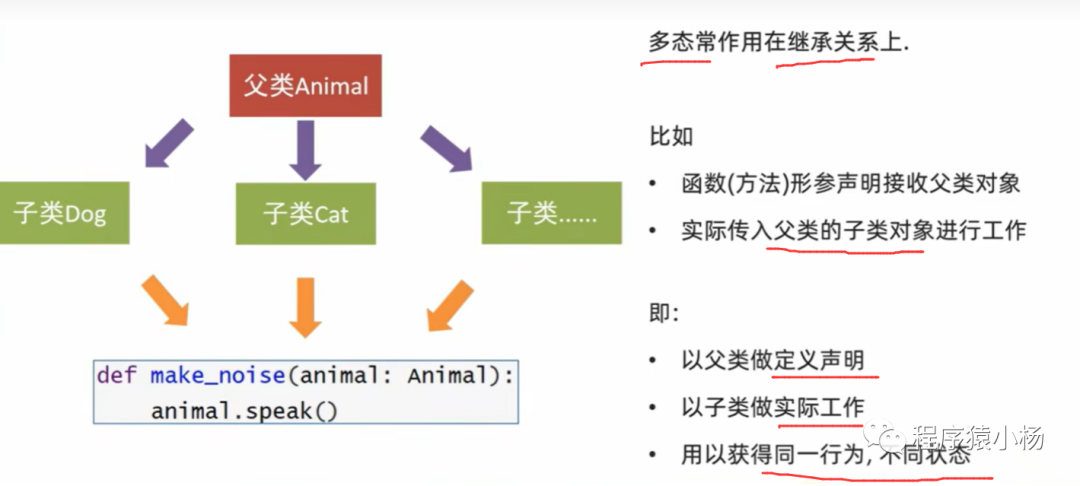
如图中所示,DeepMicroGen模型主要分为两部分,生成器(Generator)和判别器(Discriminator)。
•生成器
生成器的目标是产生更真实的伪造数据,用于填补缺失值。
•辨别器
判别器的目标是辨别真实的数据和伪造的数据,促使生成器不断地生成更加真实的虚假数据。
两者通过反复训练来相互对抗和优化,最终达到生成器生成的数据足以欺骗判别器的效果,从而提高模型的生成和预测能力。
在进入模型训练前,要对输入的微生物组数据进行预处理,使用clr方法进行了归一化,同时,为了避免某些物种在所有样本中完全缺失的情况,即数据中不会出现零值,计算了伪计数(最小相对丰度除以2)。
▷DeepMicroGen模型中的生成器(Generator)训练
先判断每个时间点上是否存在该物种,0表示数据缺失,1表示数据存在。

然后计算出时间差δf,一个向量,用于表示前一个观测值与当前时间点之间的时间差。δb,与δf定义类似,但计算方向相反,这反映了下一个观测值的时间与当前时间点之间的差异。
这些变量的计算有助于更准确地捕捉时间序列数据中的时间相关性。

接着将x(观测值)、e、δf和δb变量值输入至DeepMicroGen模型的生成器中,使用CNN模块提取捕捉系统发育关系的特征。
计算门水平的OTUs之间的Spearman相关性,得到相关性系数矩阵(一个p×p矩阵,其中p个OTUs在一个聚类中),利用下面的公式将矩阵中的每一行简化为相关系数的几何平均值,其中ρOTUjp是OTUj和OTUp之间的Spearman相关性。

然后OTUs按照几何平均值从大到小进行排序,进一步强化门水平OTU之间的关联。之后,将CNN模块分别应用于每个聚类并提取特征,该模块由两个1D-CNN层组成,卷积核大小为3,分别具有16和8个滤波器,每个滤波器后接一个Leaky ReLU激活函数和一个max-pooling层。
从每个聚类中提取的特征被连接并传递到biRNN模块。通过这种方式,DeepMicroGen模型可以同时利用门聚类和时间序列数据的相关性,提高对微生物组数据中缺失值的填充精度。
为了训练模型,需要捕捉时间序列之间的关系,以便填充缺失值。因此,在模型中增加了一层具有tanh激活函数的单层biRNN模块,用于捕捉正向和反向的时间关系。
RNN(循环神经网络)单元的定义如下:其中,Wh是权重矩阵,bh是bias,hi−1是前一个时间点的隐藏状态(hidden state)

在biRNN 模块中的生成器应用中,前向(forward)和反向(backward)RNN单元都会产生一个生成的输出,分别用 x˜f和x˜b指示。
然后,每个生成的输出将与其对应的权重系数λf和λb相乘,λf和λb是根据时间差分计算出来的,bias向量bλf和bλb是模型通过训练学习得到的参数,最终的插补值 ˜x是由输入数据和生成数据按一定比例加权组合而成,如果输入数据中存在真实值,那么生成值就会被真实值替代。
其中,1是一个全1矩阵,xe是一个矩阵,其中第i列是x乘以一个单位向量ei(即只有第i个位置是x,其他位置均为0)。

▷ DeepMicroGen模型中的判别器与生成器之间的对抗训练
判别器由一个带有LSTM单元的一层RNN模块构成,判别器的训练过程旨在最小化两个不同的损失函数lossD和lossT,以此优化判别器的性能,从而提供准确的反馈信息给生成器,改进生成器的生成结果。
其中,lossD用于评估判别器的鉴别能力,即判断一个样本是真实值还是生成值的能力;lossT用于评估判别器对时间点的预测准确性。

在生成器的训练过程中,会分别计算lossG、lossR和lossC。使得生成器产生更准确、一致且真实的填充值。
lossG表示生成器的损失函数(Generator Loss),表示生成器在生成伪造数据过程中的误差;lossR表示真实数据损失函数(Real Data Loss),表示判别器对真实数据分类的误差;lossC表示伪造数据损失函数(Fake Data Loss),表示判别器对伪造数据分类的误差。

最终,通过相互对抗和共同优化,判别器和生成器可以同时得到优化,并且生成器可以生成具有真实样本类似但又与数据集不完全相同的新数据。
DeepMicroGen是基于Tensorflow库(v1.8.0)构建的。在训练中使用了Adam优化算法,Adam的学习率被设置为0.001。
每一轮训练,生成器会在判别器经过5次迭代之后进行一次训练。当生成器的损失函数连续1000轮训练没有降低时,训练就会被结束。同时使用了Dropout(CNN层中的0.7比率)来缓解过拟合的问题。
下图为训练过程的损失函数曲线:

DeepMicroGen模型的性能评估
实验设计
使用的测试数据集包括1个模拟数据集和2个真实数据集(DIABIMMUNE和BONUS-CF数据集)。
DIABIMMUNE数据集内含从三个国家(芬兰、爱沙尼亚和俄罗斯)招募的婴儿粪便样本,这些国家在1型糖尿病和过敏的发病率方面存在很大差异。
根据16s rRNA扩增子测序,选择了133名受试者的1064份粪便样本(时间点为第4、7、10、13、16、19、22和28个月),其中包含115个种水平的OTUs。BONUS-CF数据集内含231个婴儿的粪便样本,这些婴儿中,有一部分存在囊性纤维化(CF)或高风险发展成囊性纤维化的情况。
研究人员选择了113名受试者的452份在5个月、6个月、8个月和10个月时的粪便样本,使用WGS测序。模拟数据是基于DIABIMMUNE数据集模拟了200名受试者。
使用了三种不同类型的缺失值填补方法进行性能评估。采用简单的方法(平均值和中位数)进行插补,然后比较了线性曲线拟合、三次曲线拟合和基于移动窗口(window-size = 3)的缺失值填补方法。
还使用了广泛用于纵向数据集的缺失值填补方法:基于链式方程的多重插补(MICE)和最后一次观测值向前插补(LOCF)。使用平均绝对误差(MAE)来衡量不同缺失值填补方法的性能。
★ 实验结果表明DeepMicroGen模型优于其他方法
DeepMicroGen模型在模拟数据集上的MAE为1.866,优于其他方法,其中MICE的表现次之,为1.942。其他方法的MAE分别为:三次曲线拟合(1.943)、线性曲线拟合(1.990)、Median(2.277)、Mean(2.298)、Moving window(2.452)、LOCF(2.698)。DeepMicroGen在真实数据集上也取得了最好的成绩,MAE分别为1.609和0.486。

DeepMicroGen模型利用GAN与CNN结合提取OTU特征来推算微生物组数据。研究人员为了观察不同组件对imputation性能的影响,通过移除或改变模型中的组件设计了四种变体DeepMicroGen模型。然后分别使用DIABIMMUNE数据集进行10倍交叉验证,并计算MAE进行性能评估。
如下图所示,与所有四种变体模型相比,DeepMicroGen表现出最佳性能。

评估DeepMicroGen模型在不同数据缺失率下的表现。随机(MAR)或非随机(MNAR)删除10%到80%的数据,使用DeepMicroGen模型进行缺失值填补,实验重复5次,计算MAE。结果表示除MNAR缺失率为40%外,DeepMicroGen模型在大多数情况下的MAE仍然是最低的。
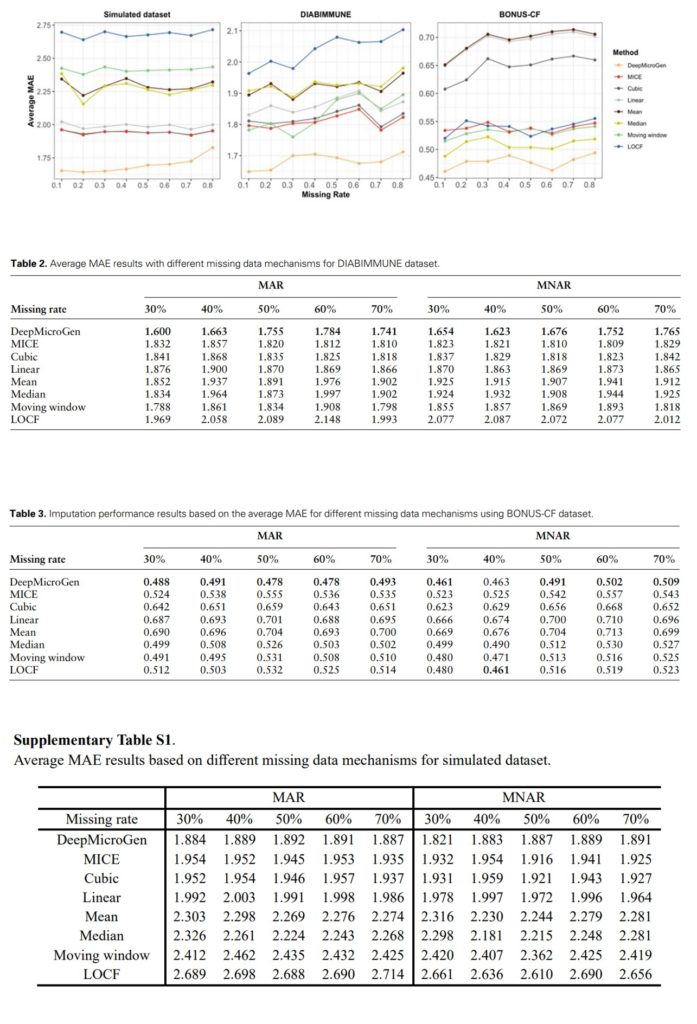
评估DeepMicroGen模型生成的插补值是否能够与原始数据保持相似性。研究人员随机丢弃10%-80%的数据,并使用DeepMicroGen和其他方法进行填补。
对于每种方法,分别比较了Shannon指数结果,NMDS分析结果。还计算量真实值和插补值分别与这两者间的pearson相关系数。
结果表示mean、median、MICE、linear和cubic方法的插补值与真实值的alpha多样性存在显著差异(P<0.05)。
MW和LOCF方法进行插补得到的数据,其alpha多样性与真实数据较为接近,但它们与真实数据的相关性较低。
★ DeepMicroGen模型的到的插补值与真实数据相似,相关性最高
而通过DeepMicroGen模型得到的插补值,其alpha多样性与真实数据相似,并且相关性最高。在比较beta多样性时,还可以看到DeepMicroGen模型生成的插补值与真实数据之间有重叠,而其他方法却几乎没有重叠。

此外,研究人员还测试了是否能够识别真实数据中相对丰度值(RA)为0的,并准确预测。准备了两个数据集合,分别是{true zero RAs}和{predicting zero RAs},分别表示真实数据集中RA=0的OTU集合和生成插补值数据集中RA=0的OTU集合。依次比较这两个集合之间的对称差,预测准确率和MAE值。

★ 能够有效保留原始数据特征,利于下游分析
这些结果都表明DeepMicroGen模型生成的插补值能够有效地保留原始数据的特征,更有助于下游分析。
评估DeepMicroGen模型是否可以提高疾病预测能力。在DIABIMMUNE数据集中,141名婴儿有鸡蛋、牛奶和花生过敏结果的临床信息,其中117名婴儿有8个时间点的所有样本,而其他25名受试者没有样本。
研究人员利用LSTM神经网络分类器对蛋、牛奶和花生过敏等疾病进行了预测,使用clr转换后的相对丰度数据作为DeepMicroGen模型数据输入,比较经重复5次5-fold交叉验证计算 的平均AUC值,与其他插补值生成方法相比,DeepMicroGen模型在所有过敏疾病的预测表现中实现了最高的改善效果,其中花生过敏的预测表现提高了19.5%。

编辑
DeepMicroGen模型的局限性
由于DeepMicroGen模型较好的结果表现是假定样本是在相同的时间间隔下生成的,如果样本的时间间隔不规律,那么DeepMicroGen模型的表现可能不太好。
因此,最好有一定数量的样本在每个时间点上。当然,对于这类数据,DeepMicroGen模型也能训练并生成插补值,只是精确度可能会降低。
结论
DeepMicroGen 模型为微生物组数据缺失值填补提供了一种有效的方法,并且在实验中表现出了良好的性能,在疾病预测方面的潜力也不逊色。
据文中介绍,基于深度学习的纵向数据填补方法已经被用于特定的研究领域,如用于阿尔茨海默病进展的MRI特征插补,患者管理的电子健康记录插补,以及肺癌风险评估的CT图像插补。
这为利用不完整的纵向数据集进行微生物组研究的科学家们提供了支持。
参考文献
Choi JM, Ji M, Watson LT, Zhang L. DeepMicroGen: a generative adversarial network based method for longitudinal microbiome data imputation. Bioinformatics. 2023 Apr 26:btad286.