数据挖掘
数据挖掘学习笔记——GEO数据库:芯片数据分析
文章目录
- 数据挖掘
- 一、芯片基础知识
- 1.1、背景
- 二、GEO数据库概述
- 2.1、基础简介
- 2.2、检索页面展示
- 三、GSE项目的三种下载方式
- 3.1、主页下载原始数据
- 3.2、主页下载表达矩阵
- 3.3、GEOquery包下载表达量
- 四、基因名与探针ID的转换技巧
- 4.1、获取对照关系
- 4.1.1、利用Bioconductor中汇总的R包
- 4.1.2、利用平台的数据
- 4.1.2.1、直接利用GEOquery包下载
- 4.1.2.2、从NCBI中下载GPL的soft包(适用网络不好时)
- 4.1.2.3、从NCBI中下载GPL的full table(适用网络不好时)
- 4.2、ID转换
- 五、差异化分析——limma(筛选差异基因)
- 5.1、简介
- 5.2、前期准备(input文件)
- 5.3、主要流程
- 5.4、结果解读
- 5.5、关于比较矩阵
- 5.6、关于分组矩阵
虽然目前市场上高通量测序成为了主流,不过芯片测序也发展了十几年,在公共数据库中留下了大量的值得被数据挖掘的数据集,所以学习一下GEO数据挖掘也是有意义的。
一、芯片基础知识
1.1、背景
-
意义:基因芯片分析就是为了通过生物信息学方法从这些芯片数据中发现可能对生物效应起作用的关键基因,从中寻找特定模式并对每个基因给予注释,从而挖掘出隐含的生物学过程并抽提出生物学的或功能层面上的意义。
-
根据芯片的使用目的,一张芯片可能包含数十、数百甚至数十万的不同序列。
- 探针:被排列成矩阵的DNA片段
- 靶标:样本RNA则被成为
-
芯片实验过程:样本mRNA首先被反转录成cDNA(在过程中同时被荧光标记),后与芯片上的核酸探针混合,互补杂交的cDNA就结合到芯片上,而未被杂交的样本被洗脱掉。芯片被一个荧光扫描仪扫描后,芯片上某个位置探针结合上了样本中互补的核酸,就在该位置显出了一个荧光点,此位置提示基因的身份,而荧光强度则提示了原始样本中该mRNA水平的高低。
-
芯片技术中的两种基本方法:单染色技术和双染色技术。
- 单染色技术:将一个样本经一种荧光标记后单独杂交的一张芯片上(使用/数据集最多)。产生的芯片数据为单通道信号数据
- 优点:方便在多张芯片之间进行比较。
- 缺点:数据变异大,需要通过重复实验来减少误差。
- 双染色技术:将两个样本用不同荧光标记后一起杂交到同一张芯片上。用于检测两种不同条件下基因表达的差异情况,如样品组与对照组。两个样本被两种不同荧光标记(Cy5(红色染料)标记,Cy3(绿色染料)标记)后,与芯片上的探针竞争杂交。产生双通道信号数据。
- 优点:此数据便于两样本间的直接比较,有助于减少数据变异性,提高组间差异表达分析的准确性,同时减少了芯片的使用量,节约了成本。
- 缺点:使用这种技术需要确定好实验设计,因而无法与其他样本进行比较。
- 单染色技术:将一个样本经一种荧光标记后单独杂交的一张芯片上(使用/数据集最多)。产生的芯片数据为单通道信号数据
-
主流平台:Affymetric公司、Agilent公司和Illumina公司。
-
数据下载:NCBI的GEO和EBI的ArrayExpress是目前最大的公开资源数据库,用于存储和发布与MIAME相容的芯片数据。
二、GEO数据库概述
2.1、基础简介
- 简介:GENE EXPRESSION OMNIBUS(GEO)数据库,是由美国国立生物技术信息中心NCBI创建并维护的基因表达数据库。起先只是为表达芯片数据准备的,后期纳入了各种NGS组学数据。这些数据包括基于单通道和双通道微阵列的实验,检测mRNA,基因组DNA和蛋白质丰度,以及非阵列技术,如基因表达系列分析(SAGE),质谱蛋白质组学数据和高通量测序数据。
- 四种基本实体类型:
- GEO Platform (GPL) 平台
- GEO Sample (GSM) 样本
- GEO Series (GSE) 系列
- GEO Dataset (GDS) 数据集
- 前三个(样本,平台和系列)由用户提供;数据集由GEO工作人员根据用户提交的数据进行编译和策划。
- 四者关联:一篇文章可以有一个或者多个GSE数据集,一个GSE里面可以有一个或者多个GSM样本。多个研究的GSM样本可以根据研究目的整合为一个GDS,不过GDS本身用的很少。而每个数据集都有着自己对应的芯片平台,就是GPL。
2.2、检索页面展示
-
页面:
-
- NCBI官网中All Databases下拉框中选择GEO DataSets
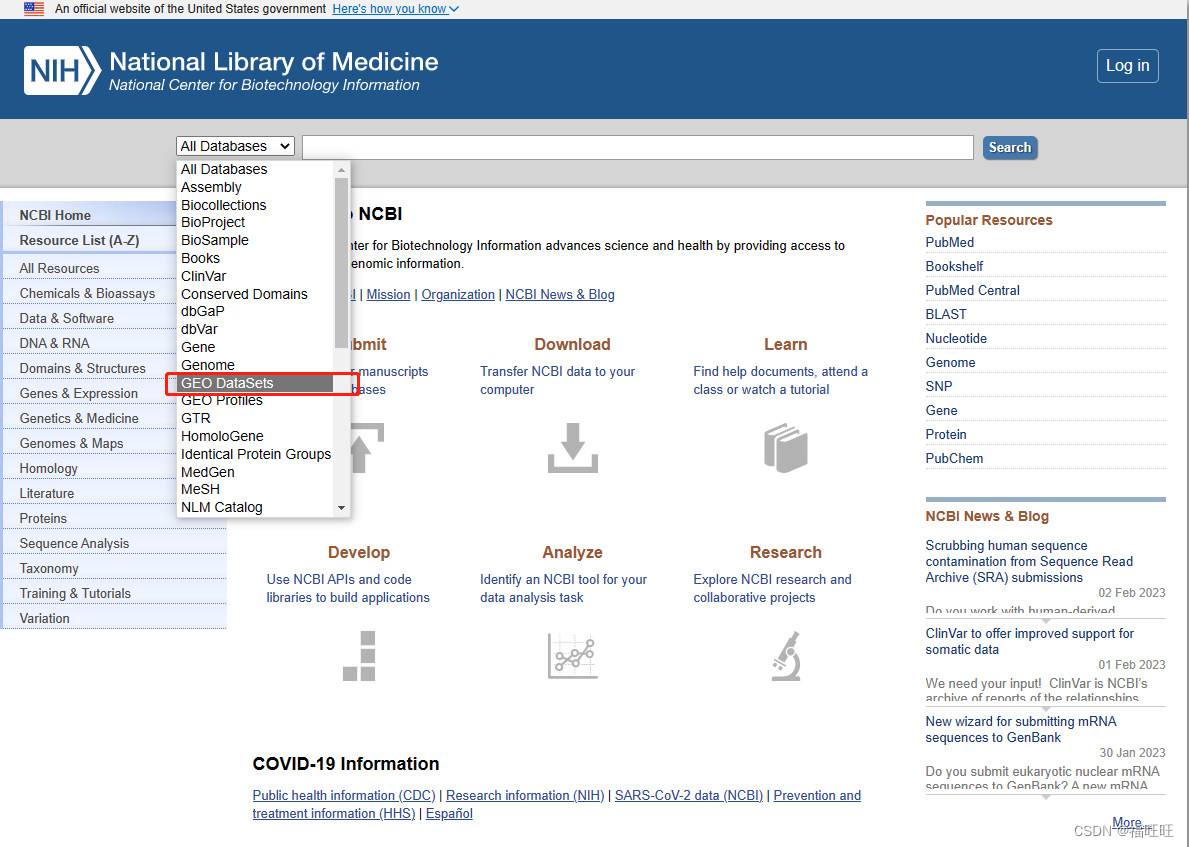
- NCBI官网中All Databases下拉框中选择GEO DataSets
-
- GEO首页
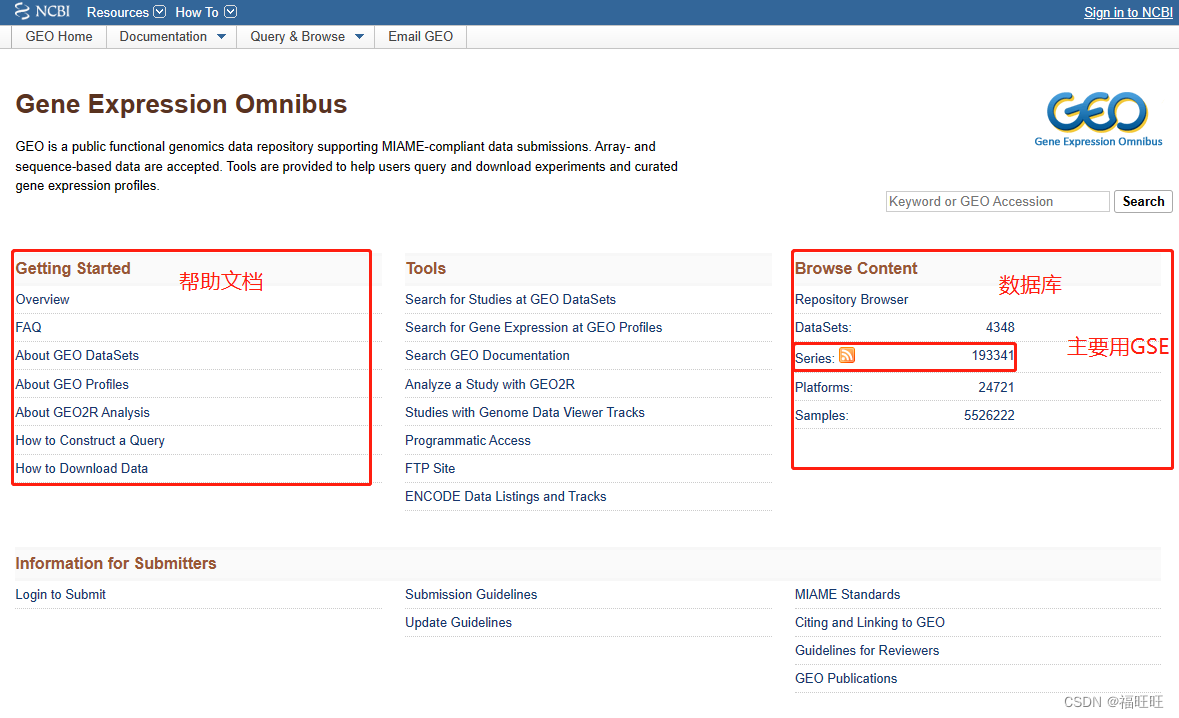
- GEO首页
-
-
检索:
-
情况一:只能确定大体研究方向(如肺癌lung cancer)
-
在NCBI官网中All Databases下拉框中选择GEO DataSets后搜索lung cancer
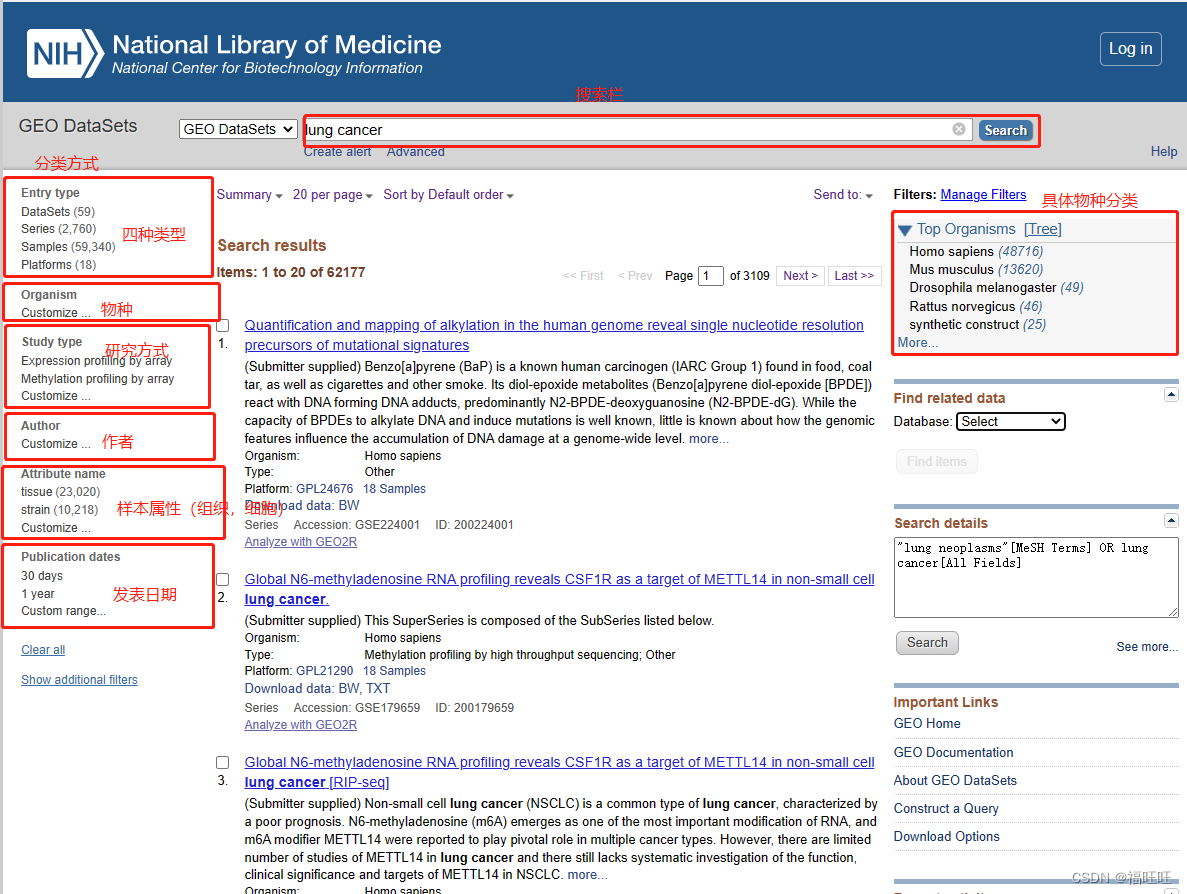
-
情况二:阅读文献后获得具体的GSE号(如GSE42872)
- 方法一:浏览器中键入https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84498(通过修改GSE号检索不同GSE)
- 方法二:GEO首页内搜索GSE号

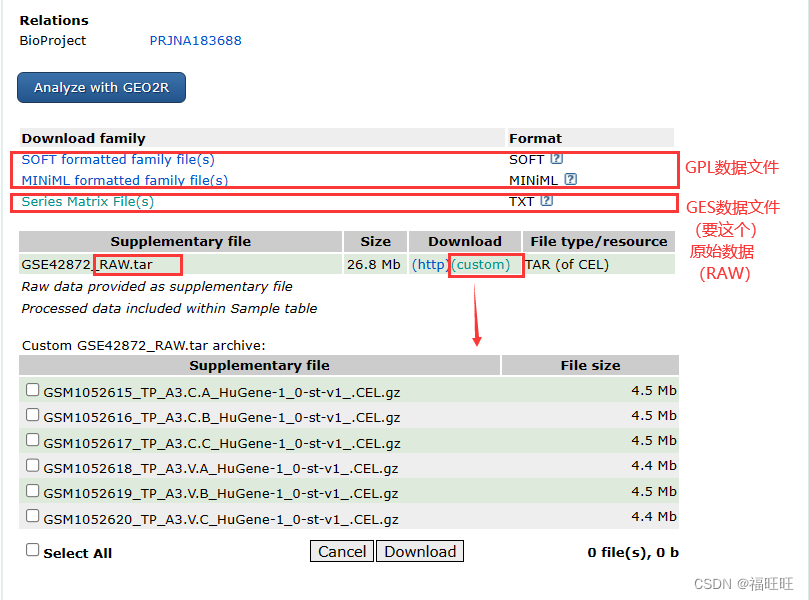
soft和miniml都是表示该platform的基础信息,比如GPL编号,上传日期等。
三、GSE项目的三种下载方式
3.1、主页下载原始数据

由于针对原始数据使用的不同芯片,分析处理的方法也不同,所以不推荐这种方法。
3.2、主页下载表达矩阵
-
下载:
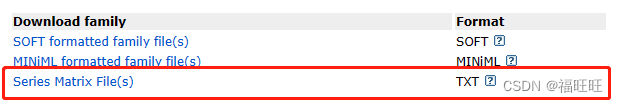
-
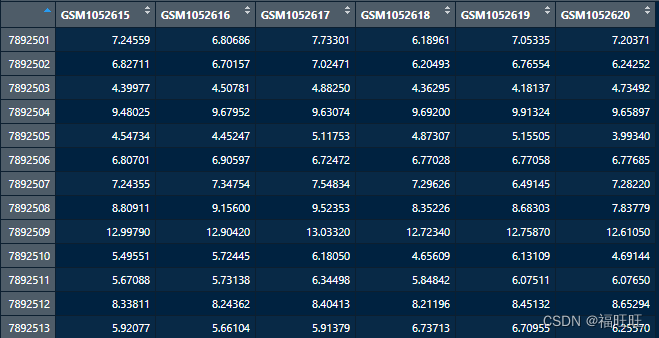
读取:
- 函数:read.table()
- 示例:
gset <- read.table("GSE42872_series_matrix.txt.gz",comment.char = "!",header = T,row.names = 1)
3.3、GEOquery包下载表达量
利用GEOquery包读取GEF号。和第二种方法的结果是一样的,都是将压缩包下载下来。
- 函数:GEOquery包的getGEO函数
- 语法:
getGEO(
GEO = NULL, # 输入GDS/GPL/GSE号
destdir = tempdir(), # 指定下载地址
GSEMatrix = TRUE, # 默认为 TRUE,表示不会进行 SOFT 格式解析
AnnotGPL = FALSE, # 表示是否使用注释 GPL 信息
getGPL = TRUE, # 是否下载 GPL 信息(SOFTT文件),指定F可以节省时间(文件大)
parseCharacteristics = TRUE # 是否解析 GSE 矩阵文件的特征信息
)
- 示例:
1、根据GDS号来下载数据,下载soft文件
gds858 <- getGEO(‘GDS858’, destdir=“.”,getGPL = F)
2、根据GPL号下载的是芯片设计的信息!
gpl96 <- getGEO(‘GPL96’, destdir=“.”,getGPL = F)
3、根据GSE号下载数据,下载series_matrix.txt.gz
gse1009 <- getGEO(‘GSE1009’, destdir=“.”,getGPL = F)
- 下载数据并获得表达矩阵
# 加载R包,suppressPackageStartupMessages函数取消了加载R包时的输出内容
suppressPackageStartupMessages(library(GEOquery))
# 下载数据,如果文件夹中有会直接读入(这边以GSE42872为例)
gset = getGEO('GSE42872', destdir=".",getGPL = F)
# 此时的gset 还是一个对象(list),需要将里面的东西取出来才能进行下一步分析。
# gset[[1]]就是list的第一层,也就是第一个GPL,由于该GSE只有一个GPL所以也只有一层list。
gset=gset[[1]]
# 获取表达矩阵
expr <- exprs(gset)

四、基因名与探针ID的转换技巧
—般分析芯片数据都需要把探针的ID切换成基因名
4.1、获取对照关系
4.1.1、利用Bioconductor中汇总的R包
我将常用的GPL平台对应的BIOconductor的R包整理为了表格,直接进行导入就可以了。
- 表格下载
# 根据GPL查询R包
platformMap <- read.csv("platformMap.csv",header = T,row.names = 1)
index <- "GPL6244" # 改GPL号
package <- platformMap$bioc_package[grep(index,platformMap$gpl)]
package_db <- paste0(package,".db")
# 下载并调用R包
BiocManager::install(package_db)
library(hugene10sttranscriptcluster.db) # 改library包
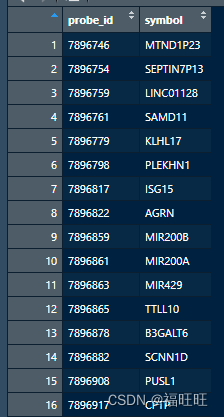
# 导出探针ID与基因名的对照矩阵
probe2symbol_df <- toTable(get(paste0(package,"SYMBOL")))
4.1.2、利用平台的数据
不是所有的平台数据都被包装为了R包,剩下的这些需要我们手动绘制ID转换表格。
4.1.2.1、直接利用GEOquery包下载
- 1、下载数据
# 下载GPL数据(也就是soft文件)
library(GEOquery)
GPL6244 <-getGEO('GPL6244',destdir =".")
# 读取对象
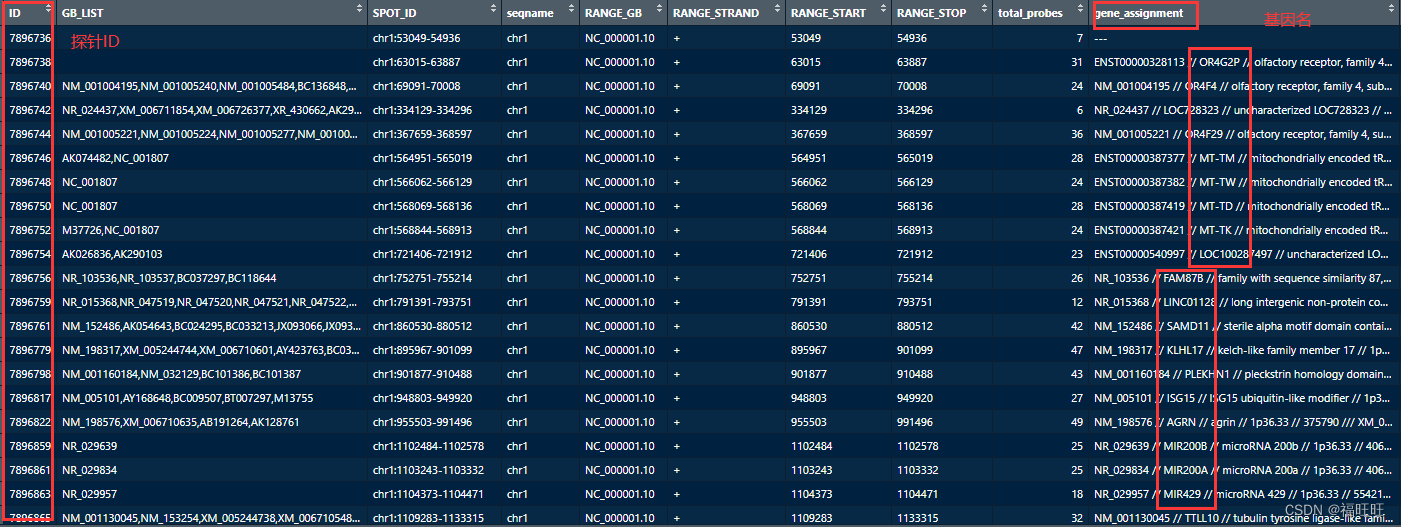
GPL6244_anno <- Table(GPL6244)
基因名称在了gene_assignment这一列的中间
- 2、提取数据,绘制为ID对照表格
我们需要将探针ID(第一列)和gene_assignment列中的基因名提取出来
library(dplyr) # 函数filter
library(tidyr) # 函数separate
probe2symbol_df1 <- GPL6244_anno %>% # 用管道进行连续操作
select(ID,gene_assignment) %>% # select函数将需要的列提取出来
filter(gene_assignment != "---") %>% # filter函数将gene_assignment列中= "---"的行全部过滤掉
separate(gene_assignment,c("drop","symbol"),sep="//") %>% # separate函数将gene_assignment按照//作为分隔符,保留前两项并命名
select(-drop)# 基因名在第二列,将第一列删除,并将结果传递给probe2symbol_df
4.1.2.2、从NCBI中下载GPL的soft包(适用网络不好时)


- 流程:
GPL6244_anno <-data.table::fread("GSE42872_family.soft.gz",skip ="ID")
# skip ="ID"不能漏
library(dplyr) # 函数filter
library(tidyr) # 函数separate
probe2symbol_df2 <- GPL6244_anno %>% # 用管道进行连续操作
select(ID,gene_assignment) %>% # select函数将需要的列提取出来
filter(gene_assignment != "---") %>% # filter函数将gene_assignment列中= "---"的行全部过滤掉
separate(gene_assignment,c("drop","symbol"),sep="//") %>% # separate函数将gene_assignment按照//作为分隔符,保留前两项并命名
select(-drop)# 基因名在第二列,将第一列删除,并将结果传递给probe2symbol_df
4.1.2.3、从NCBI中下载GPL的full table(适用网络不好时)
这里读取到的表格也可以在网页上查看

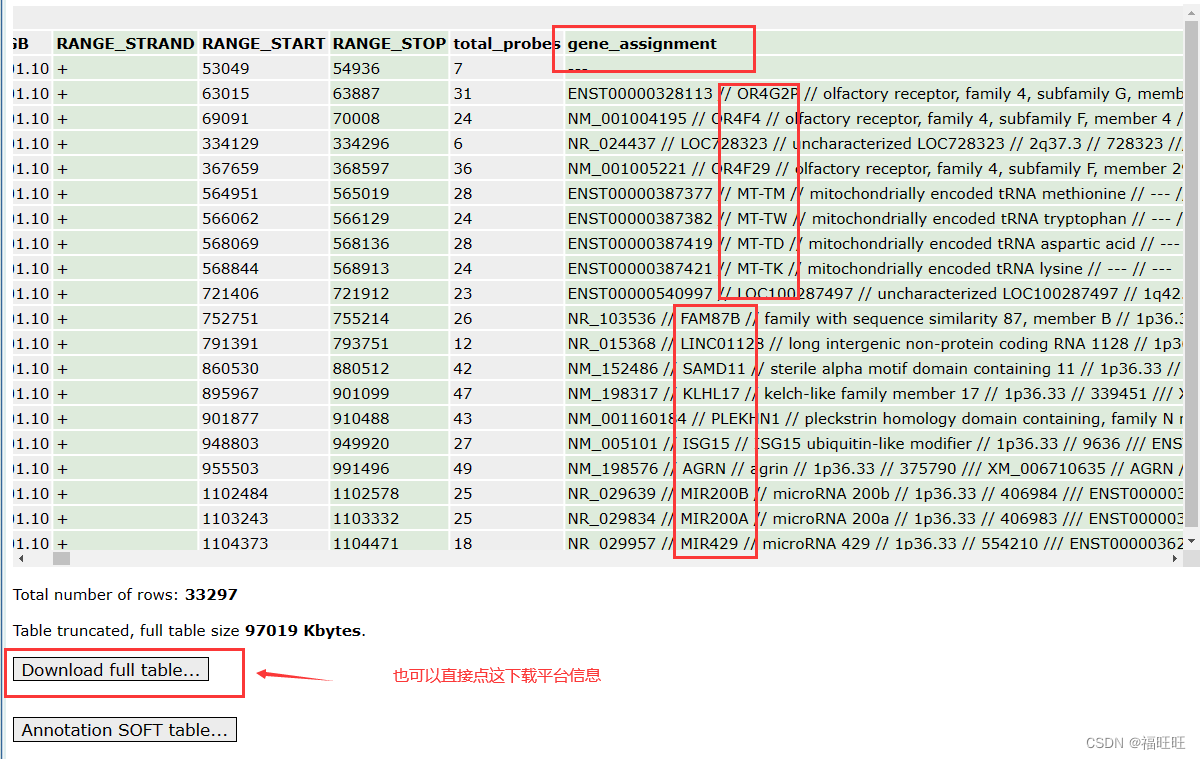
GPL6244_anno1 <-data.table::fread("GPL6244-17930.txt")
library(dplyr) # 函数filter
library(tidyr) # 函数separate
probe2symbol_df3 <- GPL6244_anno1 %>% # 用管道进行连续操作
select(ID,gene_assignment) %>% # select函数将需要的列提取出来
filter(gene_assignment != "---") %>% # filter函数将gene_assignment列中= "---"的行全部过滤掉
separate(gene_assignment,c("drop","symbol"),sep="//") %>% # separate函数将gene_assignment按照//作为分隔符,保留前两项并命名
select(-drop)# 基因名在第二列,将第一列删除,并将结果传递给probe2symbol_df
这一个例子中,利用R包导入的基因量少于平台数据整理出来的数据量

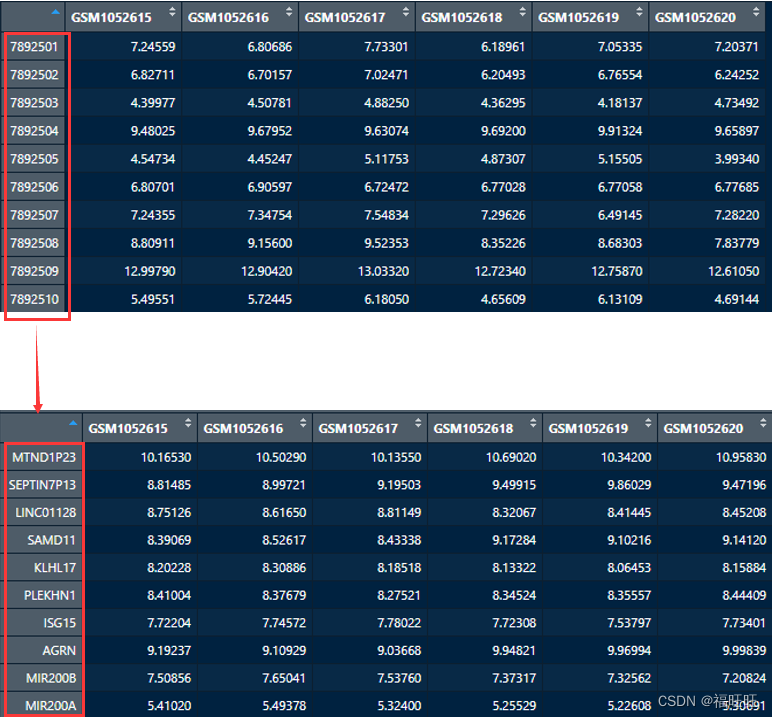
4.2、ID转换
现在我们希望将表达矩阵中的探针ID改成基因名
只需要将两个数据框按照探针名称合并就可以了, 同时在这里,我们还需要把重复的探针去掉,方法就是计算每一个探针的平均值,留下最大的那个。
# 调整两个数据框的探针名字和类型,方便合并
colnames(exprSet)[1] <- colnames(probe2symbol_df)[1]
exprSet <- as.data.frame(exprSet)
library(dplyr)
exprSet1 <- exprSet %>%
inner_join(probe2symbol_df1,by="probe_id") %>% #合并探针的信息
select(-probe_id) %>% #去掉多余信息
select(symbol, everything()) #重新排列,
#求出平均数(rowMean),并创建新的列(mutate函数)
a <- exprSet1[,c(grep("GSM", colnames(exprSet1)))]
for (i in 1:ncol(a)) {
a[,i] <- as.numeric(unlist(a[,i]))
}
a <- cbind(rowMeans(a),exprSet1)
exprSet <- a %>%
arrange(desc("rowMeans(a)")) %>% #把表达量的平均值按从大到小排序
distinct(symbol,.keep_all = T) %>% # symbol留下第一个
select(-"rowMeans(a)") %>% #反向选择去除rowMean这一列
tibble::column_to_rownames(colnames(.)[1]) # 把第一列变成行名并删除
此时得到的表达矩阵已经可以用于后续任何需要的分析了。
五、差异化分析——limma(筛选差异基因)
一般文章中会将差异基因的数量控制在200-300的范畴(当然越少越好)
这边介绍一下最适合分析芯片数据的差异化分析包——limma
5.1、简介
- 概述:Linear Models for Microarray Data(limma)基于线性模型建模

- 官方帮助文档:链接(第8章 Linear Models Overview是重点)
5.2、前期准备(input文件)
- 表达矩阵 (exprSet):芯片数据可以通过exprs() ,也就是我们上述步骤得到的表达矩阵,常规的转录组可以通过read.csv()等导入。
- 分组矩阵 (design) :将实验样本(表达矩阵的列)按照实验设计分成几组(如样本组-对照组(case - control))(实验分组与实验样本数量无关)【通过model.matrix()得到】
- 比较矩阵 (contrast):指定函数如何进行组间比较【通过makeContrasts()得到】
- 示例:
# 表达矩阵(exprSet)
# BiocManager::install('CLL')
suppressPackageStartupMessages(library(CLL))
data(sCLLex) # sCLLex是CLL包中的一个对象(例子)
sCLLex
samples=sampleNames(sCLLex)
exprSet=exprs(sCLLex)# 获取表达矩阵,列是各个样本,行是每个探针ID
pdata=pData(sCLLex)# 获取分组信息
group_list=as.character(pdata[,2])# 描述了样本的状态,需要根据它来进行样本分类
# 分组矩阵(design)
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
# 差异比较矩阵(contrast)
# 比较矩阵的组之间用-连接
contrast.matrix<-makeContrasts(paste0(unique(group_list),collapse = "-"),levels = design)
contrast.matrix ##这个矩阵声明,我们要把progres.组跟stable进行差异分析比较
# 如果分组时有三个及三个以上,进行两两对比时,比较矩阵内就需要注明对象与顺序了
# contrast.matrix <- makeContrasts(group2-group1, group3-group2, group3-group1, levels=design)
5.3、主要流程
- lmFit():线性拟合模型构建【需要:表达矩阵和分组矩阵】 ,得到的结果再和差异比较矩阵一起导入contrasts.fit()函数
- eBayes():利用上一步contrasts.fit()的结果
- topTable():利用上一步eBayes()的结果,并最终导出差异分析结果
# limma差异化分析
# step1,lmFit()
fit <- lmFit(exprSet,design)
fit2 <- contrasts.fit(fit, contrast.matrix)
# step2,eBayes()
fit2 <- eBayes(fit2)
# step3,topTable()
tempOutput = topTable(fit2, coef=1, n=Inf)
# 使用coef参数,这里设为1,也就是表示根据上面比较矩阵的第一个比较信息来提取结果,由于这个例子中只有两个分组,所以只会有一个比较信息
# n=Inf:展示的最大值设为无限大,可以不需要该参数
# 参数adjust="BH"表示对p值的校正方法,包括了:"none", "BH", "BY" and "holm",默认为BH,适用与多数情况,所以这边没有修改这个参数。
nrDEG = na.omit(tempOutput) # 删除NA值
write.csv(nrDEG,"C:/Users/16748/Desktop/limma_notrend.results.csv",quote = F) # 输出差异分析矩阵
- P值校正:p值是针对单次检验,假设采用的p值为小于0.05,我们通常认为这个基因在两组样本中的表达是有显著差异的,但是仍旧有5%的概率表示这个基因并不是差异基因。但是,当两组样本中有20000个基因采用同样的检验方式进行统计检验时,就会遇到一个问题:单次犯错的概率为0.05, 如果进行20000次检验,那么就会有0.05*20000=1000 个基因在组间的差异被错误估计。简单来说,P值校正是为了降低大样本中假阳性的数量,提升数据可信度。
5.4、结果解读
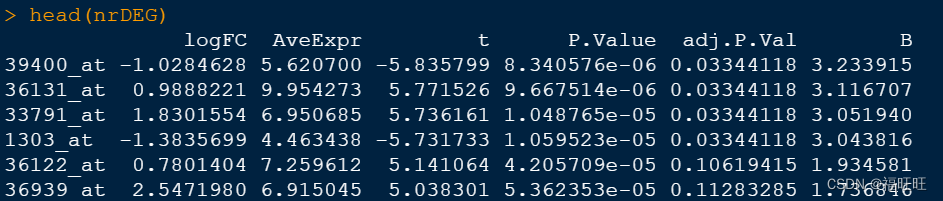
内容包括AveExpr值(所有样本的平均表达量)、logFC值(差异倍数)、t值、P值、q值(即adj.P.Val值)和B值。一般logFC值、P值、q值和AveExpr值用来作为差异表达的判断标准,比如差异倍数在2倍以上、平均表达在10以上、P值小于0.01等。
t值是统计量,
• 第一列:“logFC”是两组表达值之间以2为底对数化的的变化倍数(Fold change, FC),由于基因表达矩阵本身已经取了对数,这里实际上只是两组基因表达值均值之差;
• 第二列:“AveExpr”是该探针组所在所有样品中的平均表达值;
• 第三列:“t”是贝叶斯调整后的两组表达值间 T 检验中的 t 值,根据t值可以判断p值进而判断模型是否显著;
• 第四列:“P.Value”是贝叶斯检验得到的 P 值(越小越好);
• 第五列:“adj.P.Val”是调整后的 P 值(也称为q值,越小越好).
• 第六列:“B”是经验贝叶斯得到的标准差的对数化值。
5.5、关于比较矩阵
上述提到差异分析需要三个矩阵,表达矩阵包含样本信息,而样本必须进行分组,才能分析,所以这两个矩阵肯定需要,但比较矩阵则不一定需要。
先看下示例
- 不需要差异比较矩阵:
library(CLL)
data(sCLLex)
library(limma)
design <- model.matrix(~factor(sCLLex$Disease))
fit <- lmFit(sCLLex,design)
fit <- eBayes(fit)
topTable(fit,coef=2,adjust='BH')
# 结果
logFC AveExpr t P.Value adj.P.Val B
39400_at 1.0285 5.621 5.836 8.341e-06 0.03344 3.234
36131_at -0.9888 9.954 -5.772 9.668e-06 0.03344 3.117
33791_at -1.8302 6.951 -5.736 1.049e-05 0.03344 3.052
1303_at 1.3836 4.463 5.732 1.060e-05 0.03344 3.044
36122_at -0.7801 7.260 -5.141 4.206e-05 0.10619 1.935
36939_at -2.5472 6.915 -5.038 5.362e-05 0.11283 1.737
41398_at 0.5187 7.602 4.879 7.824e-05 0.11520 1.428
32599_at 0.8544 5.746 4.859 8.207e-05 0.11520 1.389
36129_at 0.9161 8.209 4.859 8.212e-05 0.11520 1.389
37636_at -1.6868 5.697 -4.804 9.355e-05 0.11811 1.282
- 需要差异比较矩阵:
library(CLL)
data(sCLLex)
library(limma)
design <- model.matrix(~0+factor(sCLLex$Disease))
colnames(design) <- c('progres.','stable')
fit <- lmFit(sCLLex,design)
cont.matrix <- makeContrasts('stable-progres.',levels = design)
fit2 <- contrasts.fit(fit,cont.matrix)
fit2 <- eBayes(fit2)
topTable(fit2,adjust='BH')
# 结果
logFC AveExpr t P.Value adj.P.Val B
39400_at 1.0285 5.621 5.836 8.341e-06 0.03344 3.234
36131_at -0.9888 9.954 -5.772 9.668e-06 0.03344 3.117
33791_at -1.8302 6.951 -5.736 1.049e-05 0.03344 3.052
1303_at 1.3836 4.463 5.732 1.060e-05 0.03344 3.044
36122_at -0.7801 7.260 -5.141 4.206e-05 0.10619 1.935
36939_at -2.5472 6.915 -5.038 5.362e-05 0.11283 1.737
41398_at 0.5187 7.602 4.879 7.824e-05 0.11520 1.428
32599_at 0.8544 5.746 4.859 8.207e-05 0.11520 1.389
36129_at 0.9161 8.209 4.859 8.212e-05 0.11520 1.389
37636_at -1.6868 5.697 -4.804 9.355e-05 0.11811 1.282
- 解析:差异比较矩阵的需要与否,主要看分组矩阵如何制作的
- design=model.matrix(~factor(sCLLex$Disease))
- design=model.matrix(~0+factor(sCLLex$Disease))
- 第一种方法把需要比较的组做出到了一列,需要比较多次,就有多少列,第一列是截距不需要考虑,第二列开始往后用coef这个参数可以把差异分析结果一个个提取出来。
- 第二种方法,仅仅是分组而已,组之间需要如何比较,需要自己再制作差异比较矩阵,通过makeContrasts函数来控制如何比较。
- 个人看法:用第二种方法(需要比较矩阵)
- 第一种方法在算法上优于第二种,在构建矩阵时其将常数项都列为1,在运算时就可以忽略常数项(关于线性方程的算法,可以参考下这篇文章 )
- 但是第二种方法可以自行定义组间比较的方式,其更加方便。第一种方法由于是写死的,固定为矩阵中的“1”表示的样本减去矩阵中的“0”表示的样本,其logFC与t值可能出现符号相反值的情况(由原先的正表达变为负表达了)
5.6、关于分组矩阵
差异分析的一个使用难点就是构建分组矩阵。具体使用方法在帮助文档的第九章(p42)都有讲解,这边就不过多赘述了。