ChemBioServer 是一个提供高级化学化合物过滤、聚类和网络分析的服务器,旨在支持药物发现和药物再利用(drug repurposing)。它集成了多种工具和网络服务,以便更高效地筛选、分析和可视化化学化合物。
网站地址:
https://chembioserver.vi-seem.eu/index.php
其主要包括以下功能:
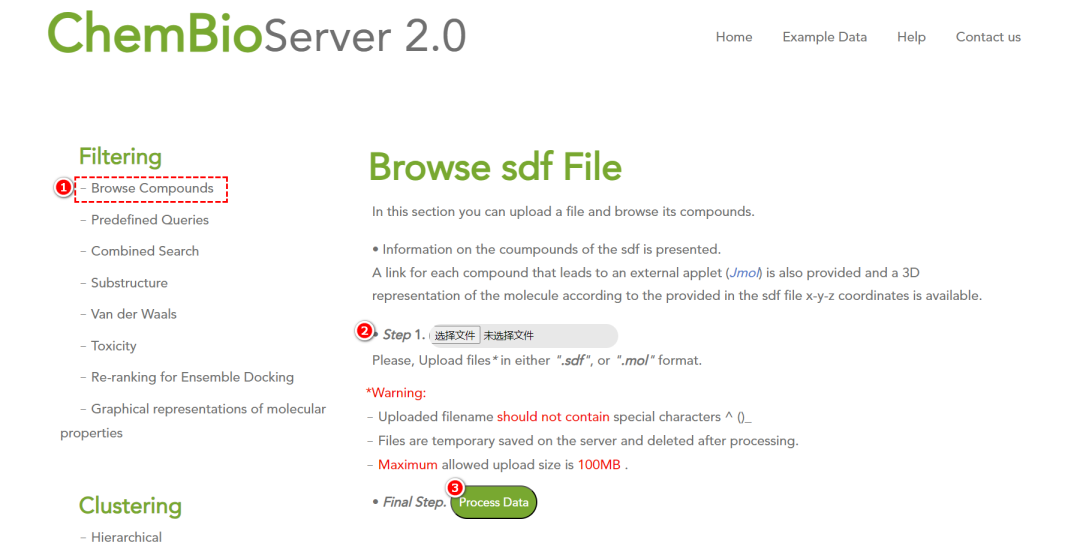
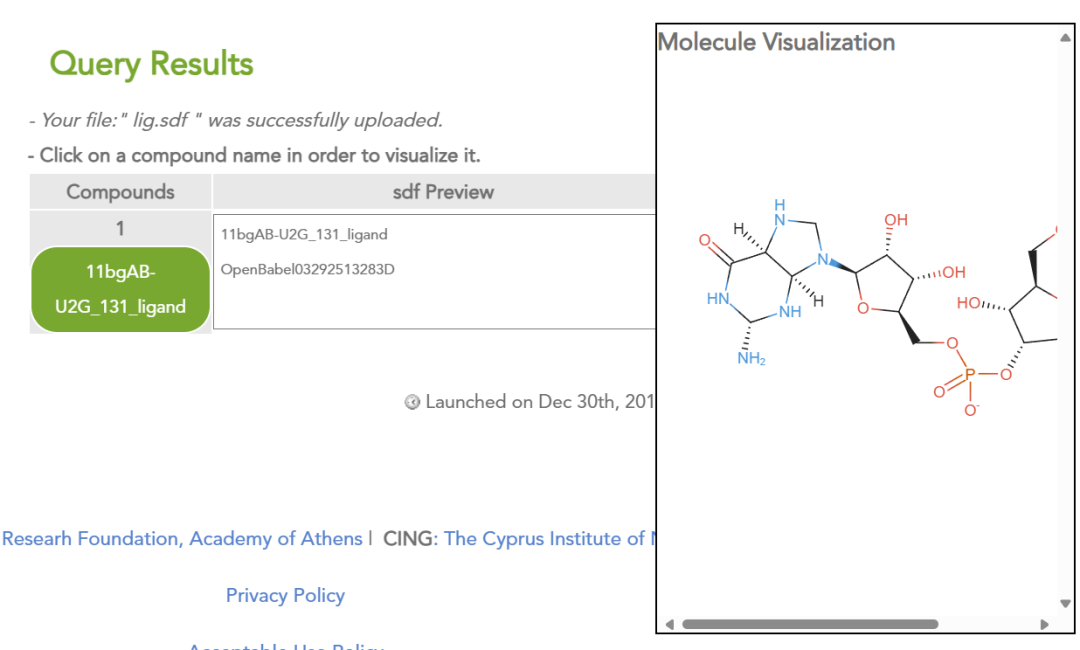
1. 化合物上传与浏览
-
• 支持 2D 形式浏览化合物结构(仅支持sdf或mol格式的文件)。
2. 化合物筛选
-
• 基于理化性质过滤:筛选符合药物可成药性(如 Lipinski 规则)的化合物。

-
• 基于子结构的筛选:使用自定义 .sdf 文件过滤含特定子结构的化合物。
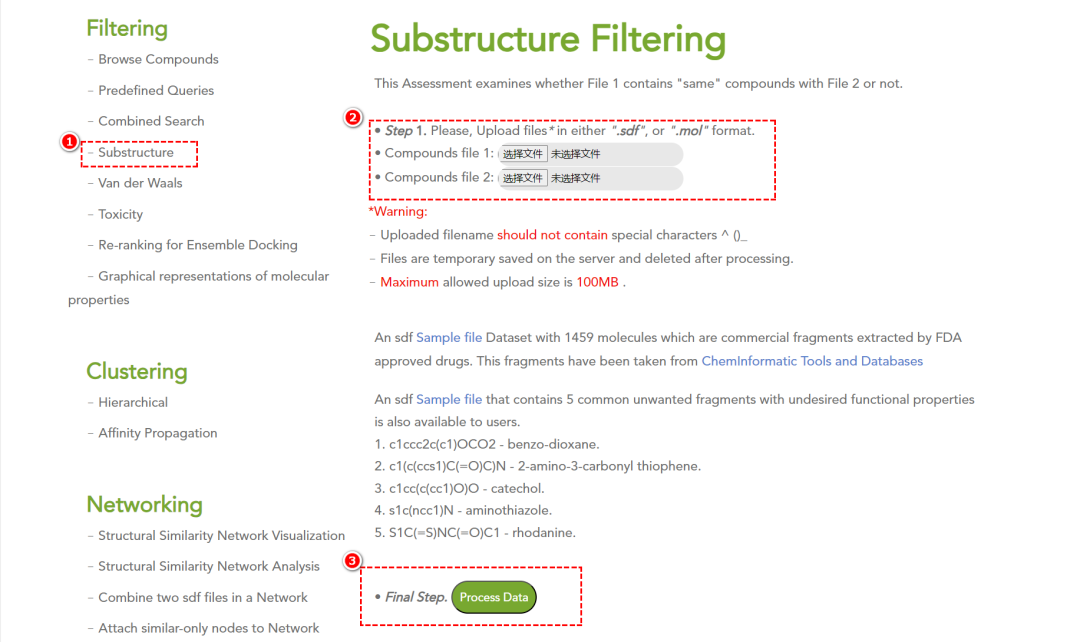
-
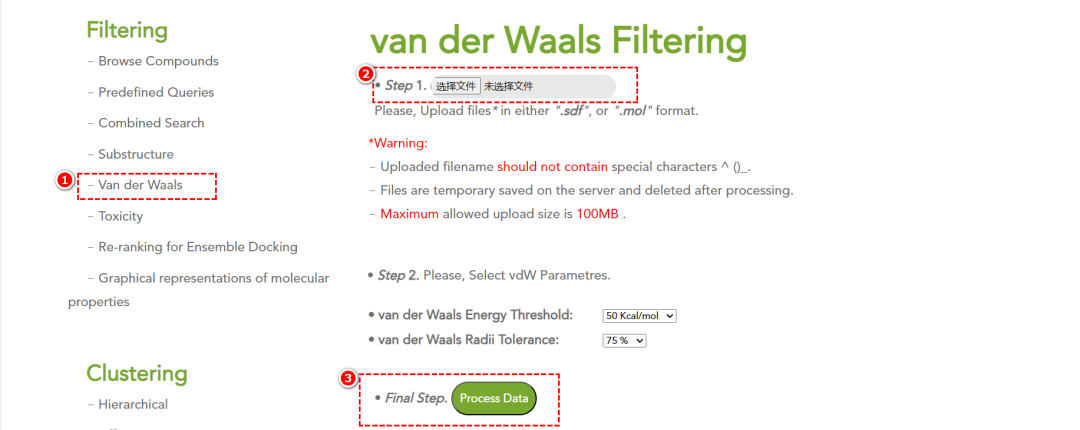
• 范德华过滤:通过距离和能量测试排除不符合标准的分子。
-
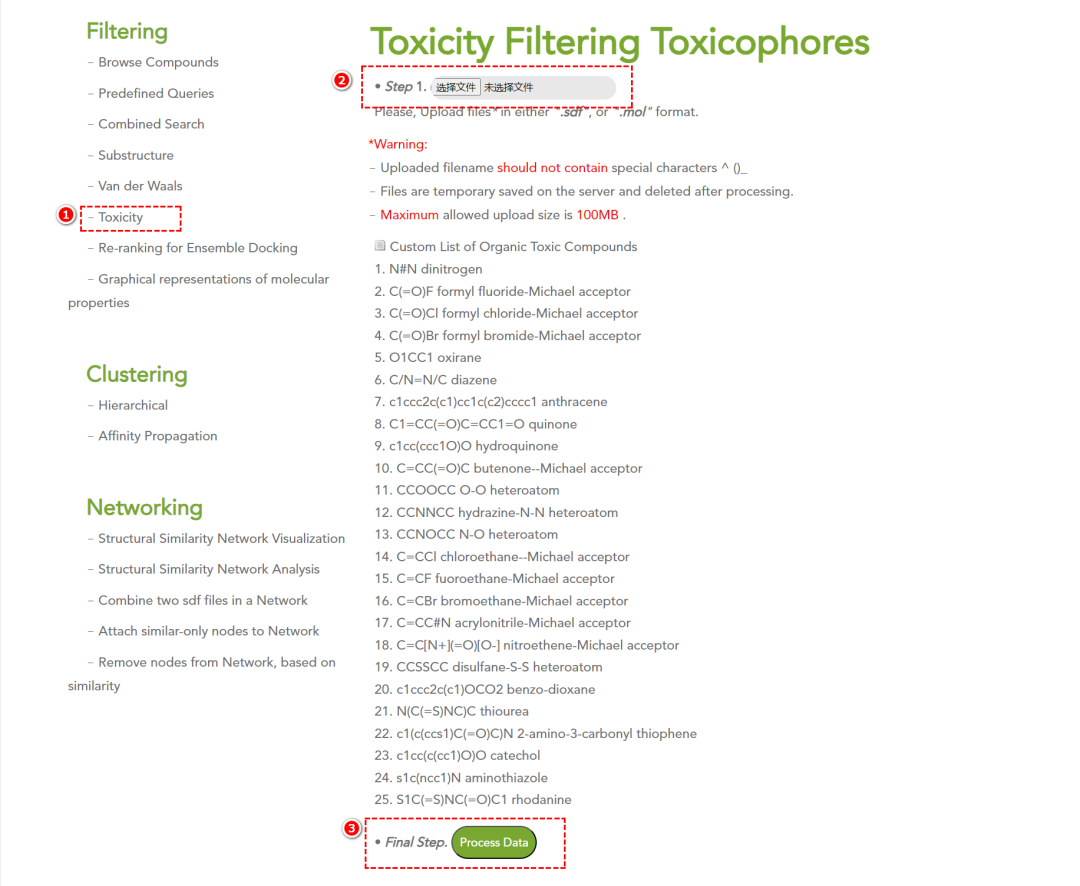
• 毒性筛选:基于有机毒性根(toxic organic roots)检测潜在毒性化合物。
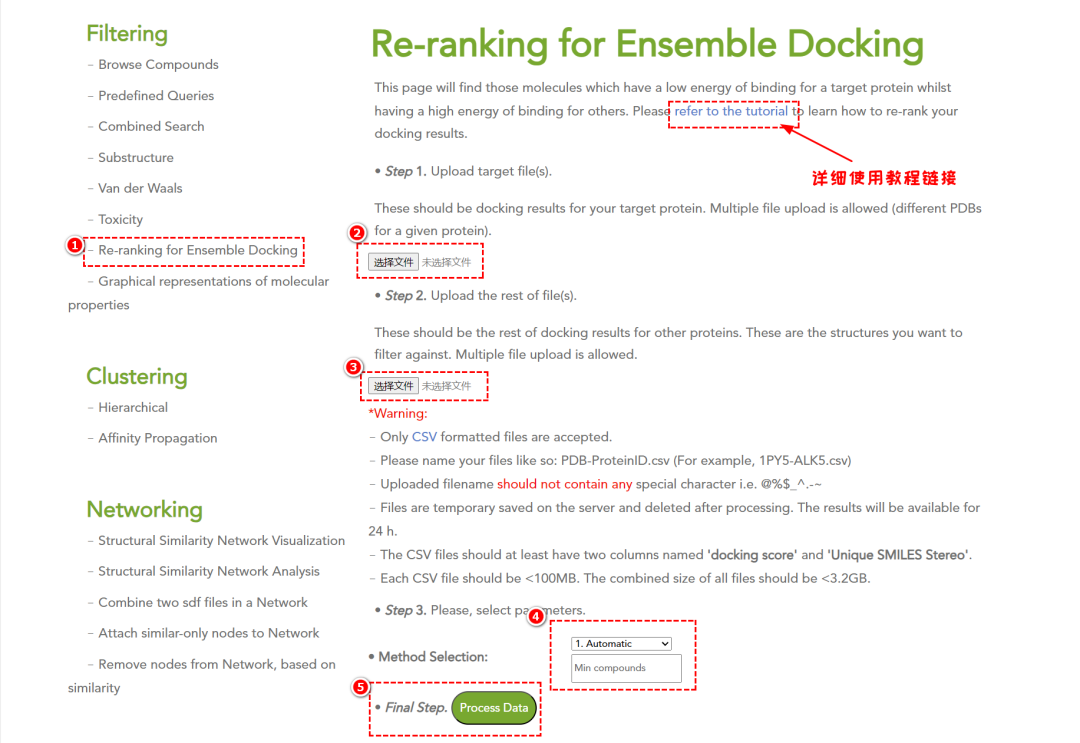
3. 交叉分子对接(Cross-Docking)
预测化合物与靶标蛋白的结合情况,支持交叉对接分析。
此功能将查找那些与目标蛋白结合能量较低而与其他蛋白结合能量较高的分子。
4. 化合物聚类分析
-
• 层次聚类(Hierarchical Clustering):支持 4 种不同的距离度量方式。
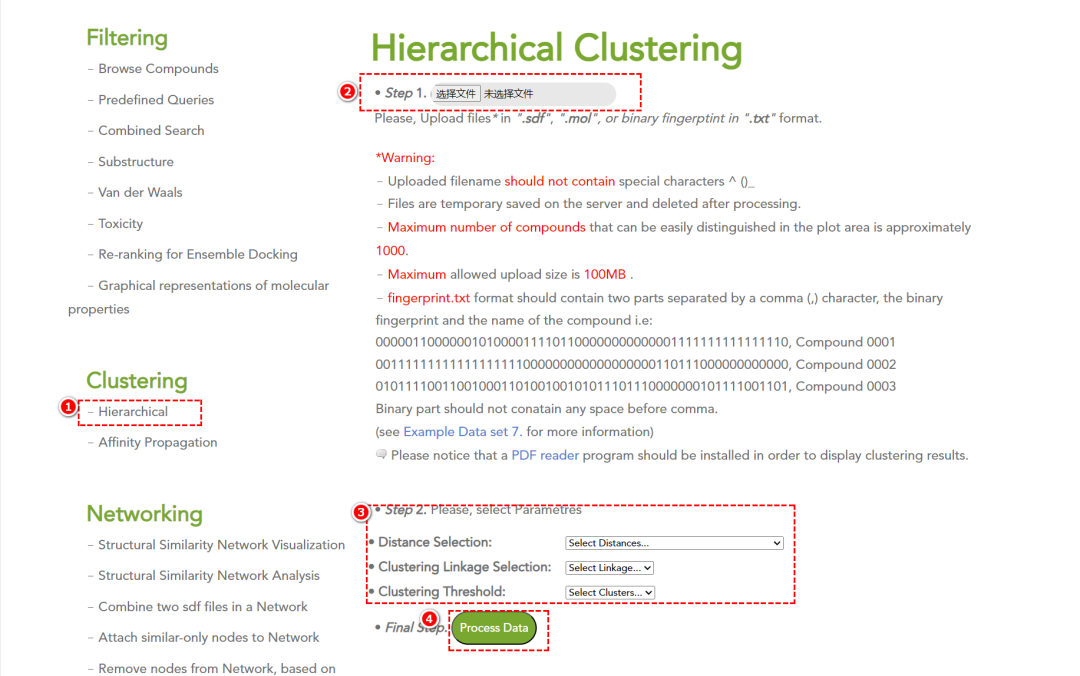
-
• Affinity Propagation 聚类:自动识别代表性化合物(exemplars),提高筛选效率。
Affinity Propagation(亲和传播)算法以化合物指纹(compound fingerprints)之间的成对相似性作为输入,将它们视为潜在的代表性化合物(exemplars)。该算法通过数据点之间的信息传递进行聚类,并不断优化,直到收敛到最优解。最终,每个聚类的代表性化合物(exemplars)被确定,并推荐给研究人员进行进一步分析。
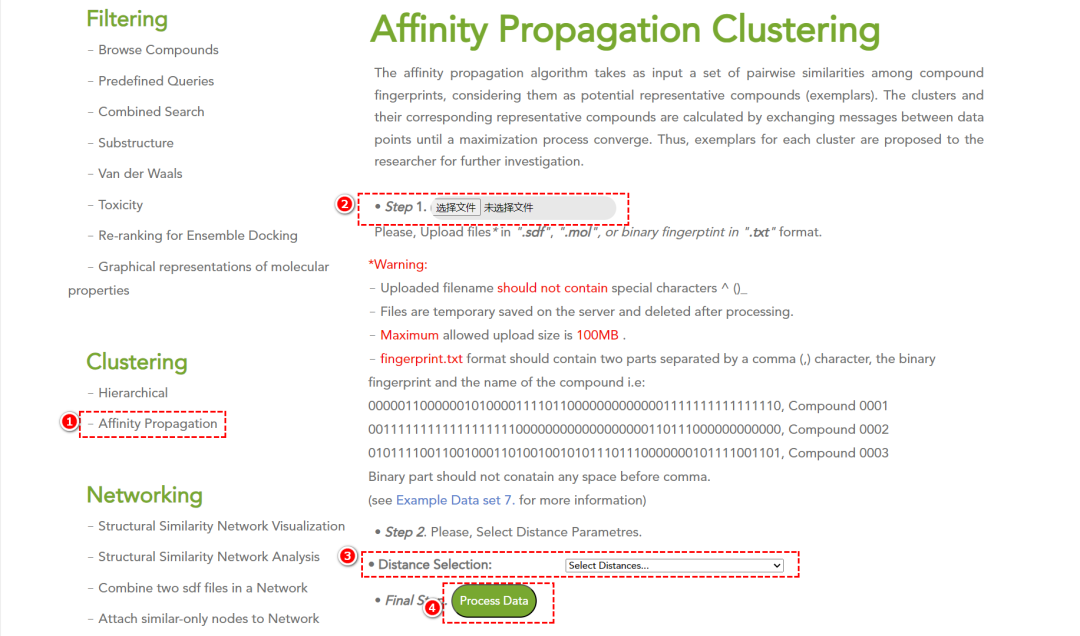
5. 结构相似性网络分析
-
• 结构相似性网络(SSN)分析:揭示化学库内部的结构模式,有助于发现潜在的活性分子。
该功能是使用Rcpi 包中的 calcDrugFPSim 计算得到的相似性矩阵被用作邻接矩阵(adjacency matrix),并利用 igraph 包构建图(graph)。通过网络分析(network analysis),执行后会生成一个表格,展示各节点的网络度量(node metrics)。
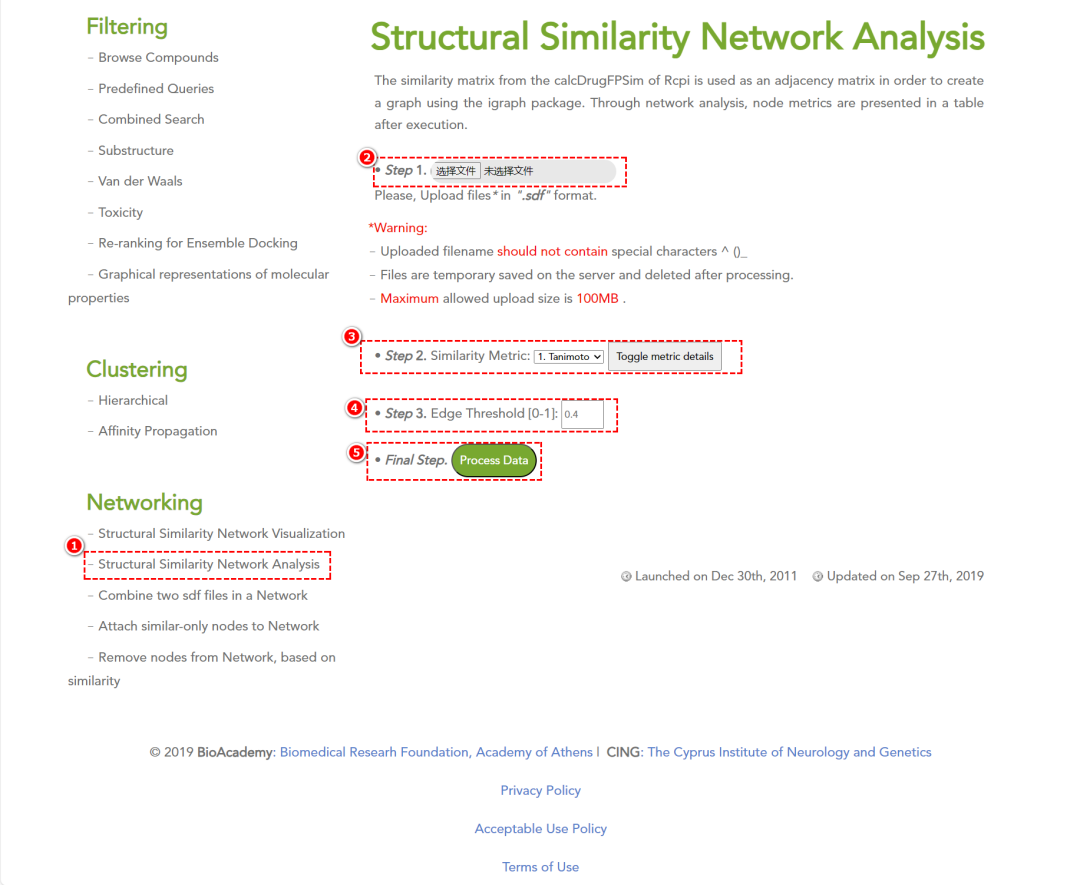
-
• 结构相似性网络(SSN)可视化:直观展示化合物之间的相似性关系。
化合物的结构相似性网络(Structural Similarity Network)是通过药物相似性矩阵构建的,该矩阵由 Rcpi 包中的 calcDrugFPSim 函数计算,基于 分子指纹(molecular fingerprints) 来评估药物分子的相似性。在构建网络时,用户需要选择相似性度量(Similarity Metric),并设定边的阈值(cutoff threshold),该阈值取值范围为 [0,1],用于决定哪些化合物之间应该绘制连接。最终,网络图(graph)使用vis.js进行可视化呈现。

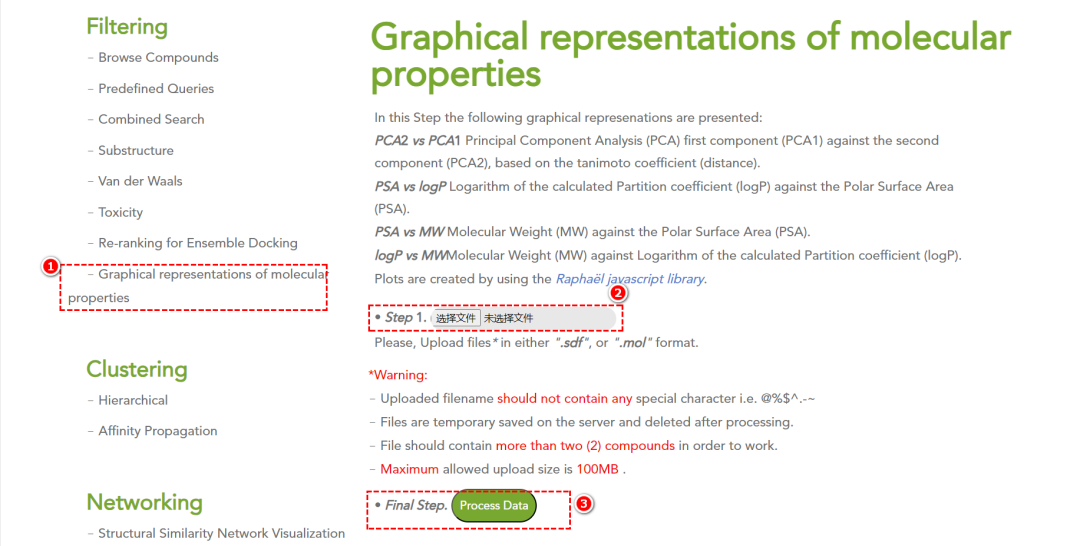
6. 化合物属性可视化
在此步骤中,将呈现以下图形表示:
-
•
PCA2 vs PCA1
主成分分析(PCA):第一主成分(PCA1)与第二主成分(PCA2)的散点图。
相似性度量基于Tanimoto 系数(Tanimoto coefficient)(即距离计算)。
-
•
PSA vs logP
极性表面积(PSA, Polar Surface Area) 与 分配系数的对数值(logP) 之间的关系图。
-
•
PSA vs MW
分子量(MW, Molecular Weight) 与 极性表面积(PSA) 的散点图。
-
•
logP vs MW
分子量(MW) 与 分配系数的对数值(logP) 之间的关系图。