1.sh plink 格式转化
plink1.map/plink1.ped ------plink2.bim/fam/bed
plink --file 1001genomes_snps_only_ACGTN1 --make-bed --out plink2
2.sh 群体结构分析
时间过久:
for K in 2 3 4 5 6 7 8 9 10;
do admixture --cv plink2.bed
K
∣
t
e
e
a
d
m
i
x
t
r
u
e
K | tee admixtrue
K∣teeadmixtrue{K}.out; done
这里你要有一个k值, 如果你不知道你的群体能分为几个类群, 可以做一个测试, 比如从1~7分别分群, 然后看他们的cv值哪个小, 用那个k值.
短时间:
已知有3类可以做群体结构分析
admixture hapmap3.bed 3
我们的格式:
只有三列Q结果
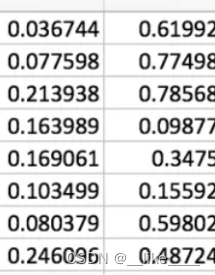
调整群体结构文件的格式:(以下为正常格式)
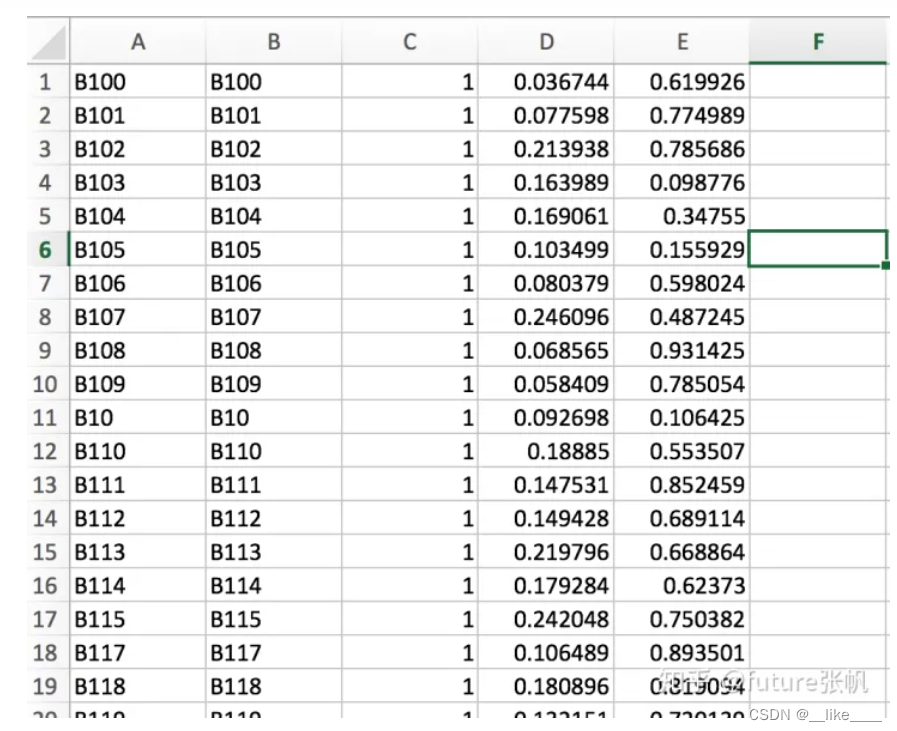
生成plink2.3.Q
调整格式:
使用plink1.ped第一列替换plink2.3.Q的第四列
awk ‘FNR==NR{a[NR]=$1;next}{$4=a[FNR]}1’ plink1.ped plink2.3.Q >tmp.txt
使用使用plink1.ped第一列替换tmp.txt的第五列
awk ‘FNR==NR{a[NR]=$1;next}{$5=a[FNR]}1’ plink1.ped tmp.txt >tmp1.txt
交换顺序tmp1.txt
awk ‘{print $5,$4,$1,$2,$3 > “tmp1.txt”}’ tmp1.txt
echo 1>tmp1.txt
第三列增加一列1
awk ‘{print $1,$2,1,$3,$4,$5}’ tmp1.txt > emmax.cov.txt
3.加入群体结构为协变量进行全基因组关联分析
8.sh
#!/bin/bash
#SBATCH -p v6_384
#SBATCH -N 1
#SBATCH -n 1
#SBATCH -c 4
export PATH=/public3/home/sch7166/emmax/emmax-beta-07Mar2010:$PATH
emmax -v -d 10 -t 1001genomes_snps_maf0.1 -p final.txt -k 1001genomes_snps_maf0.1.hIBS.kinf -c emmax.cov.txt -o adimixtrue_var
生成adimixtrue_var.ps文件