作为一个刚刚开始进行单细胞转录组分析的菜鸟,R语言底子没有,有时候除了会copy外,如果你让我写个for循环,我只能cross my fingers。。。。
于是我看见了https://satijalab.org/seurat/,Seurat是一个R软件包,设计用于QC、分析和探索单细胞RNA-seq数据。Seurat旨在帮助用户能够识别和解释单细胞转录组学中的的异质性来源,并通过整合各种类型的单细胞数据,能够在单个细胞层面上进行系统分析。
里面非常详细的介绍了这个单细胞转录组测序的workflow,包括添加了很多的其他功能,如细胞周期 (Seurat亮点之细胞周期评分和回归)等。
但里面有蛮多的代码的原理其实我并不太清楚 (读完这个,还不懂,来找我,重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)),这次我就介绍一下里面让我曾经困惑的几个问题以及比较nice的解答!(令我万万没想到,找答案比自己写答案确实困难的多。。。)
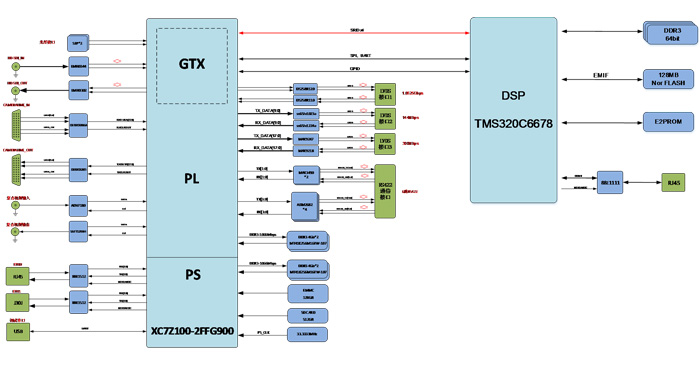
1. 有关merge函数的问题
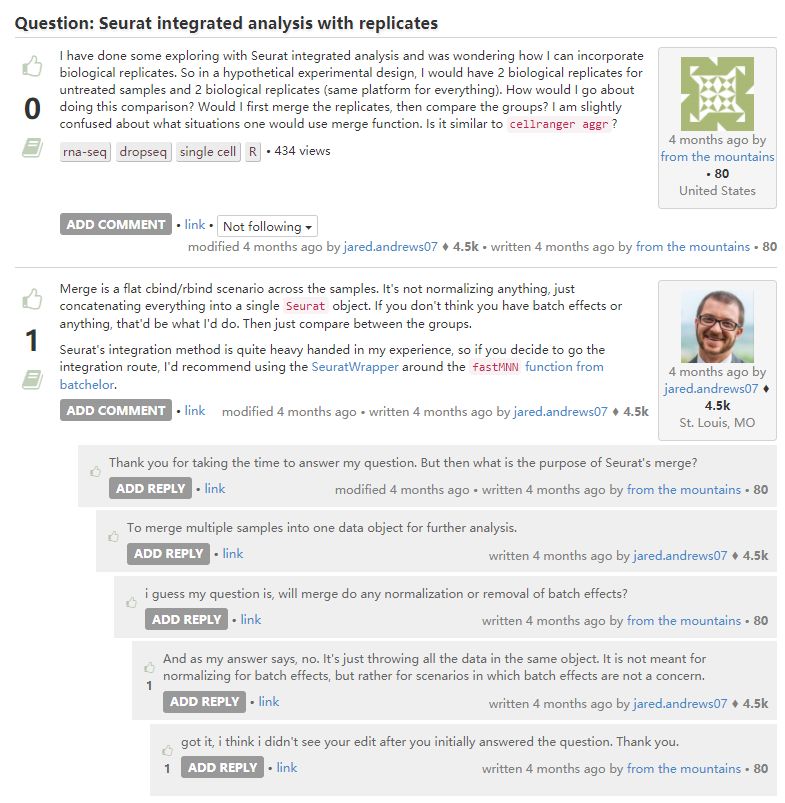
merge只是放在一起,fastMNN才是真正的整合分析。
2. 有关PC的选择
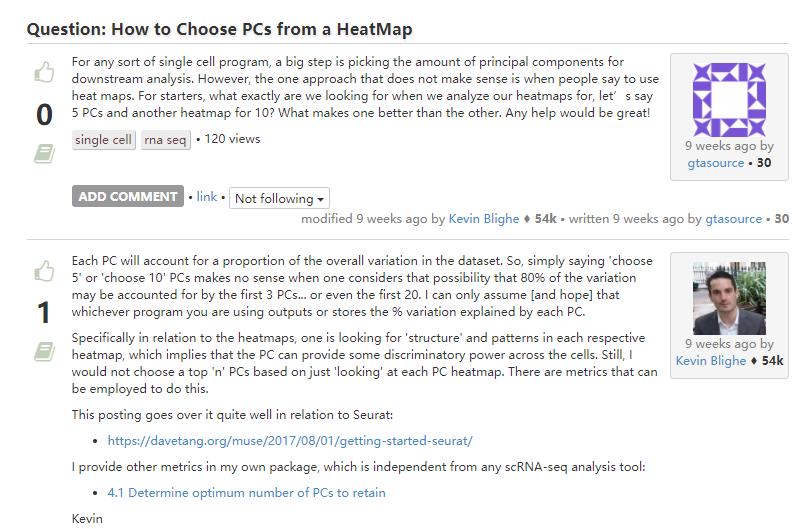
-
Seurat应用
JackStraw随机抽样构建一个特征基因与主成分相关性值的背景分布,选择富集特征基因相关性显著的主成分用于后续分析。对大的数据集,这一步计算会比较慢,有时也可能不会找到合适的临界点。 -
建议通过
ElbowPlot来选,找到拐点或使得所选PC包含足够大的variation了 (80%以上),便合适。然后再可以在这个数目上下都选几个值试试,最好测试的时候往下游测试些,越下游越好,看看对结果是否有影响。 -
另外一个方式可以是根据与各个主成分相关的基因的GSEA功能富集分析选择合适的主成分 (一文掌握GSEA,超详细教程)。
-
一般选择
7-12都合适。实际分析时,可以尝试选择不同的值如10,15或所有主成分,结果通常差别不大。但如果选择的主成分比较少如5等,结果可能会有一些变化。 -
另外要注意:用了这么多年的PCA可视化竟然是错的!!!
3. 细胞周期相关基因
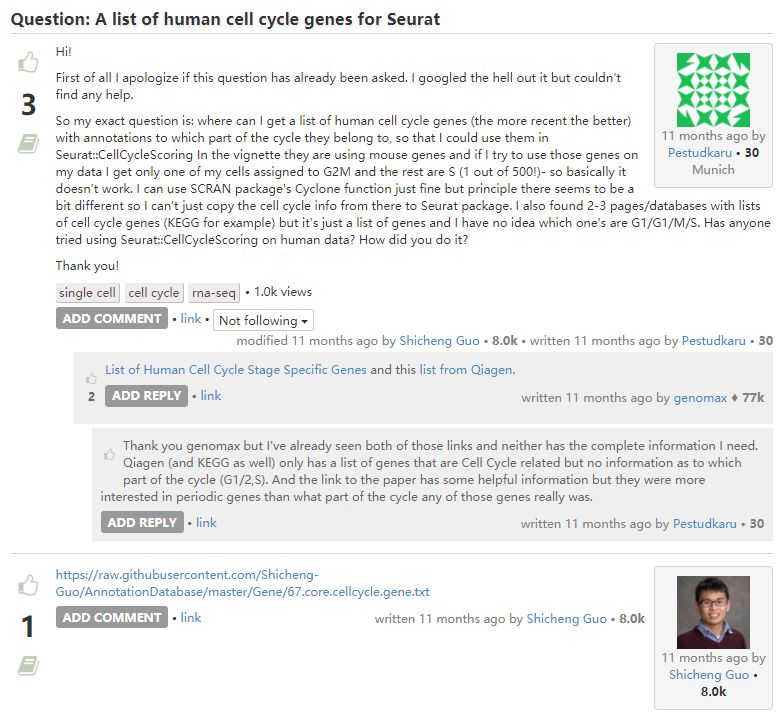
Pairs of genes (A, B) are identified from a training data set where in each pair, the fraction of cells in phase G1 with expression of A > B (based on expression values in training.data) and the fraction with B > A in each other phase exceeds fraction. These pairs are defined as the marker pairs for G1. This is repeated for each phase to obtain a separate marker pair set.
4. Seurat对象导入Monocle
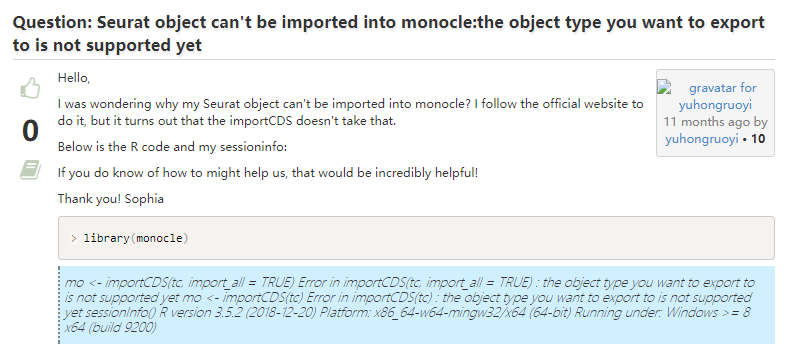
其实这个问题我也遇到了,并且已经有人给出了解决方案。(生信宝典注:官方最开始没支持Seurat3,我写了个简易的,在https://github.com/cole-trapnell-lab/monocle-release/issues/311,现在应该更新全面支持Seurat3了。)
seurat_data <- newimport(SeuratObject)
###重新定义了import函数,称为newimport
newimport <- function(otherCDS, import_all = FALSE) {
if(class(otherCDS)[1] == 'Seurat') {
requireNamespace("Seurat")
data <- otherCDS@assays$RNA@counts
if(class(data) == "data.frame") {
data <- as(as.matrix(data), "sparseMatrix")
}
pd <- tryCatch( {
pd <- new("AnnotatedDataFrame", data = otherCDS@meta.data)
pd
},
#warning = function(w) { },
error = function(e) {
pData <- data.frame(cell_id = colnames(data), row.names = colnames(data))
pd <- new("AnnotatedDataFrame", data = pData)
message("This Seurat object doesn't provide any meta data");
pd
})
# remove filtered cells from Seurat
if(length(setdiff(colnames(data), rownames(pd))) > 0) {
data <- data[, rownames(pd)]
}
fData <- data.frame(gene_short_name = row.names(data), row.names = row.names(data))
fd <- new("AnnotatedDataFrame", data = fData)
lowerDetectionLimit <- 0
if(all(data == floor(data))) {
expressionFamily <- negbinomial.size()
} else if(any(data < 0)){
expressionFamily <- uninormal()
} else {
expressionFamily <- tobit()
}
valid_data <- data[, row.names(pd)]
monocle_cds <- newCellDataSet(data,
phenoData = pd,
featureData = fd,
lowerDetectionLimit=lowerDetectionLimit,
expressionFamily=expressionFamily)
if(import_all) {
if("Monocle" %in% names(otherCDS@misc)) {
otherCDS@misc$Monocle@auxClusteringData$seurat <- NULL
otherCDS@misc$Monocle@auxClusteringData$scran <- NULL
monocle_cds <- otherCDS@misc$Monocle
mist_list <- otherCDS
} else {
# mist_list <- list(ident = ident)
mist_list <- otherCDS
}
} else {
mist_list <- list()
}
if(1==1) {
var.genes <- setOrderingFilter(monocle_cds, otherCDS@assays$RNA@var.features)
}
monocle_cds@auxClusteringData$seurat <- mist_list
} else if (class(otherCDS)[1] == 'SCESet') {
requireNamespace("scater")
message('Converting the exprs data in log scale back to original scale ...')
data <- 2^otherCDS@assayData$exprs - otherCDS@logExprsOffset
fd <- otherCDS@featureData
pd <- otherCDS@phenoData
experimentData = otherCDS@experimentData
if("is.expr" %in% slotNames(otherCDS))
lowerDetectionLimit <- otherCDS@is.expr
else
lowerDetectionLimit <- 1
if(all(data == floor(data))) {
expressionFamily <- negbinomial.size()
} else if(any(data < 0)){
expressionFamily <- uninormal()
} else {
expressionFamily <- tobit()
}
if(import_all) {
# mist_list <- list(iotherCDS@sc3,
# otherCDS@reducedDimension)
mist_list <- otherCDS
} else {
mist_list <- list()
}
monocle_cds <- newCellDataSet(data,
phenoData = pd,
featureData = fd,
lowerDetectionLimit=lowerDetectionLimit,
expressionFamily=expressionFamily)
# monocle_cds@auxClusteringData$sc3 <- otherCDS@sc3
# monocle_cds@auxOrderingData$scran <- mist_list
monocle_cds@auxOrderingData$scran <- mist_list
} else {
stop('the object type you want to export to is not supported yet')
}
return(monocle_cds)
}5. 不同条件下画热图
-
R语言 - 热图绘制 (heatmap)
-
R语言 - 热图简化
-
R语言 - 热图美化
-
利用ComplexHeatmap绘制热图(一)
-
获取pheatmap聚类后和标准化后的结果
6. Seurat对象转化为.h5ad对象
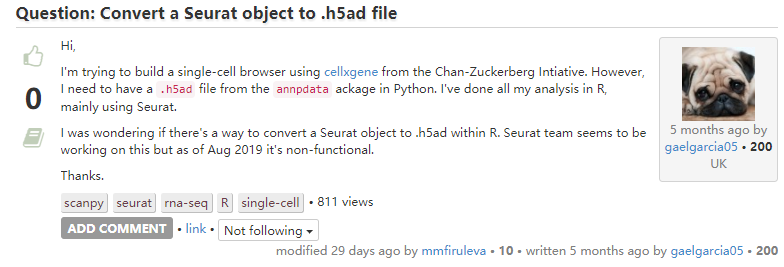
Answer:Looks like the way to do it is to write to loom format via loomR(https://github.com/mojaveazure/loomR), then read that into anndata (https://anndata.readthedocs.io/en/latest/api.html) to be written as an .h5ad file.
Single cell folks need to a pick a file format/structure and stick with it.
生信宝典注:不同文件类型和格式之间的转换确实是一个问题,好在易生信提供了好的解决方案。来这试试?易生信线下课推迟,线上课程免费看
7. 根据基因取细胞子集

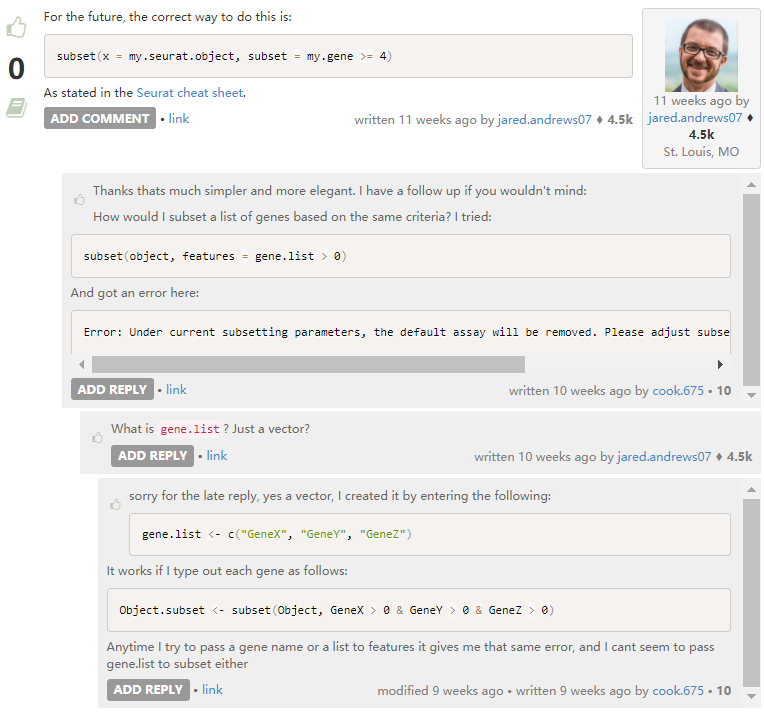
也可以提取特定Cluster,进行进一步细分。
# 提取特定cluster,继续后续分析。
ident_df <- data.frame(cell=names(Idents(pbmc)), cluster=Idents(pbmc))
pbmc_subcluster <- subset(pbmc, cells=as.vector(ident_df[ident_df$cluster=="Naive CD4 T",1]))8. 筛选条件的设置
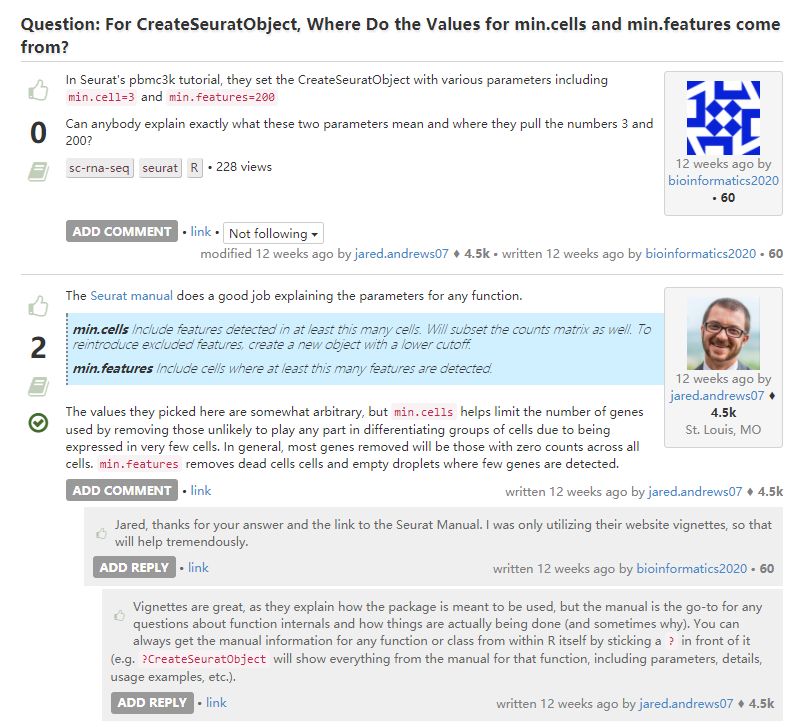
这是最开始读入的初始设置,后面质控还有更多筛选方式。
质控是用于保证下游分析时数据质量足够好。由于不能先验地确定什么是足够好的数据质量,所以只能基于下游分析结果(例如,细胞簇注释)来对其进行判断。因此,在分析数据时可能需要反复调整参数进行质量控制 (生信宝典注:反复分析,多次分析,是做生信的基本要求。这也是为啥需要上易生信培训班而不是单纯依赖公司的分析。)。从宽松的QC阈值开始并研究这些阈值的影响,然后再执行更严格的QC,通常是有益的。这种方法对于细胞种类混杂的数据集特别有用,在这些数据集中,特定细胞类型或特定细胞状态可能会被误解为低质量的异常细胞。在低质量数据集中,可能需要严格的质量控制阈值。具体见:
-
对一篇单细胞RNA综述的评述:细胞和基因质控参数的选择
-
重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)
陷阱和建议:
通过可视化检测到的基因数量、总分子数和线粒体基因的表达比例的分布中的异常峰来执行QC。
联合考虑这3个变量,而不是单独考虑一个变量。
尽可能设置宽松的QC阈值;
如果下游聚类无法解释时再重新设定严格的QC阈值。
如果样品之间的QC变量分布不同(存在多个强峰),则需要考虑样品质量差异,应按照
Plasschaert et al. (2018)的方法为每个样品分别确定QC阈值。
9. RunTSNE不是在聚类

区分好聚类 (FindClusters)和降维 (PCA,tSNE,UMAP)。
-
聚类是直接基于距离矩阵的经典无监督机器学习问题。
通过最小化簇内距离或在降维后的表达空间中鉴定密集区域,将细胞分配给簇。
方法有层级聚类、K-menas、基于图的聚类等 (基因共表达聚类分析及可视化,基因表达聚类分析之初探SOM)。
-
tSNE和UMAP目前主要用于可视化。用于可视化的降维必然涉及信息丢失并改变细胞之间的距离。因此tSNE/UMAP图应仅只用于解释或传达基于更精确的、更多维度的定量分析结果。这样可以保证分析充分利用了压缩到二维空间时丢失的信息。假如二维图上呈现的细胞分布与使用更多数目的PC进行聚类获得的结果之间存在差异,应倾向于相信后者(聚类)的结果。(如何使用Bioconductor进行单细胞分析?)
-
还在用PCA降维?快学学大牛最爱的t-SNE算法吧, 附Python/R代码
10. Seurat与线粒体基因
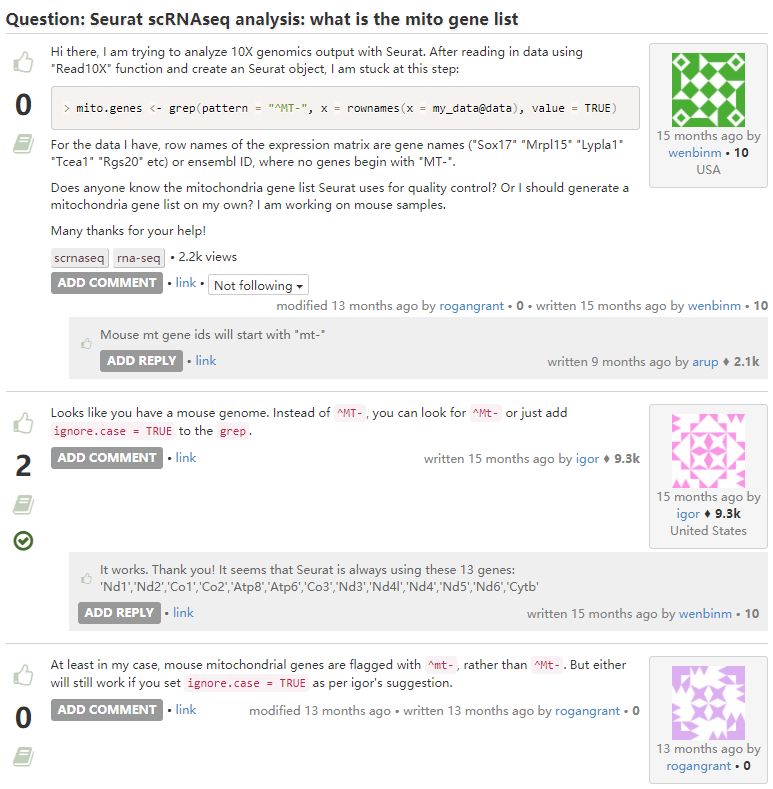
这是一个简单的操作问题,读懂代码就好解决了。送您一句话:Some years back when I was less experienced not being sure if I did an analysis right used to keep me up at night … I am still not sure if I do it right now but I sleep better ;-)

图源:Giphy
参考来源
-
Biostar: https://www.biostars.org/t/seurat/?page=4&sort=update&limit=all%20time&q=
撰文/Tiger
编辑/生信宝典