介绍
RNA-seq 目前是测量细胞反应的最突出的方法之一。RNA-seq 不仅能够分析样本之间基因表达的差异,还可以发现新的亚型并分析 SNP 变异。本教程[1]将涵盖处理和分析差异基因表达数据的基本工作流程,旨在提供设置环境和运行比对工具的通用方法。请注意,它并不适用于所有类型的分析,比对工具也不适用于所有分析。此外,本教程的重点是给出一般的分析流程。对于更大规模的研究,强烈建议使用集群来增加内存和计算能力。由于完整版过长,因此分为两部分,需要获取完整版的,请跳转文末。
项目配置
安装conda
Miniconda 是一个全面且易于使用的 Python 包管理器。 Miniconda 旨在将您当前的 Python 安装替换为具有更多功能且模块化的 Python ,因此您可以删除它而不会损坏您的系统。它不仅允许您安装 Python 包,还可以创建虚拟环境并访问大型生物信息学数据库。
# 下载 macOS
wget https://repo.continuum.io/miniconda/Miniconda3-latest-MacOSX-x86_64.sh -O ~/miniconda.sh
# 下载 LINUX
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda.sh
# 安装
bash miniconda.sh -b -f -p ~/miniconda
# 添加环境变量
echo 'PATH="$HOME/miniconda/bin:$PATH' >> ~/.bash_profile
# 更新环境变量
source ~/.bash_profile
# 为 conda 添加下载源
conda config --add channels conda-forge
conda config --add channels defaults
conda config --add channels r
conda config --add channels bioconda
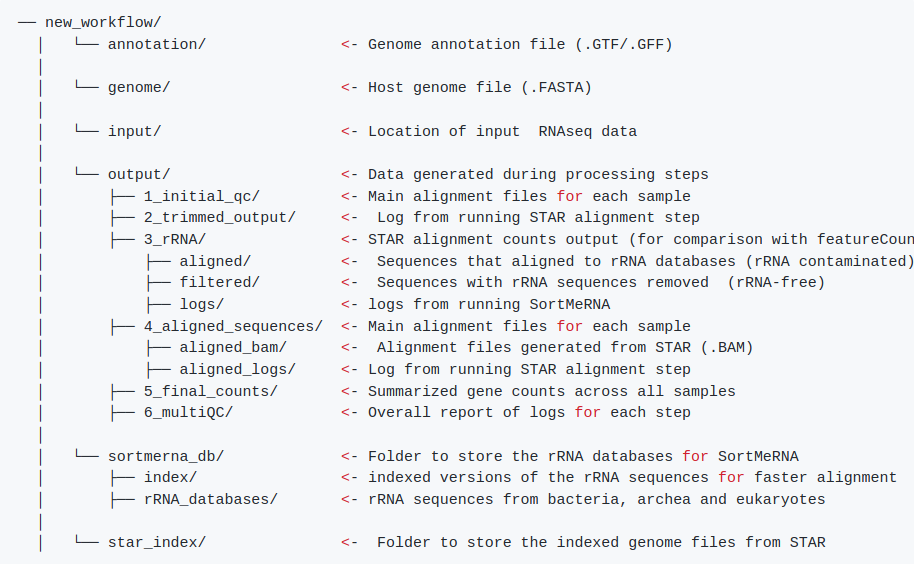
项目结构
项目结构组织适当是可重复研究的关键。在处理和分析期间,会创建许多文件。为了最好地组织并提高分析的可重复性,最好使用简单的文件结构。直观的结构允许其他研究人员和合作者按照步骤进行操作。本教程中的结构只是组织数据的一种方式,您可以选择您最习惯的方式。
-
clone(无法使用git的读者,请后台回复 " rna " )
# Clone
git clone https://github.com/twbattaglia/RNAseq-workflow new_workflow
# 进入目录
cd new_workflow # 完整结构如下图
基因组下载
要查找差异表达基因或异构体转录本,您首先需要一个参考基因组进行比较。对于任何比对,我们需要 .fasta 格式的基因组,还需要 .GTF/.GFF 格式的注释文件,它将基因组中的坐标与带注释的基因标识符相关联。这两个文件都是执行比对和生成计数矩阵所必需的。请注意,不同数据库(Ensembl、UCSC、RefSeq、Gencode)具有相同物种基因组的不同版本,并且注释文件不能混合。在本流程中,将使用 Gencode 的基因组。
-
小鼠 (Gencode)
# 基因组文件下载
wget -P genome/ ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_mouse/release_M12/GRCm38.p5.genome.fa.gz
# 注释文件下载
wget -P annotation/ ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_mouse/release_M12/gencode.vM12.annotation.gtf.gz
# 解压
gunzip genome/GRCm38.p4.genome.fa.gz
gunzip annotation/gencode.vM12.annotation.gtf.gz
-
人 (Gencode)
# 基因组文件下载
wget -p genome/ ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_25/GRCh38.p7.genome.fa.gz
# 注释文件下载
wget -P annotation/ ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_25/gencode.v25.annotation.gtf.gz
# 解压
gunzip genome/GRCh38.p7.genome.fa.gz
gunzip annotation/gencode.v25.annotation.gtf.gz
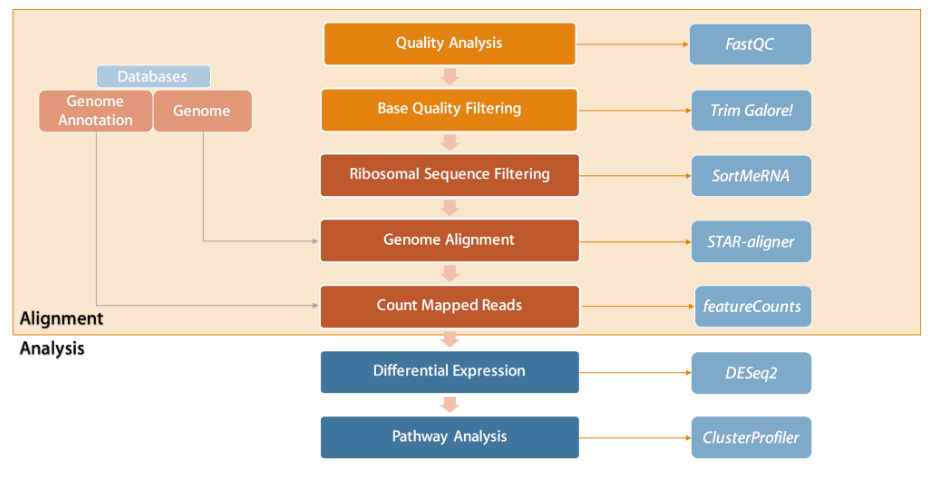
流程
示例数据:如果您想使用示例数据来练习,请运行以下命令下载老鼠的 RNAseq 数据。
wget -P input/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/001/SRR1374921/SRR1374921.fastq.gz
wget -P input/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/002/SRR1374922/SRR1374922.fastq.gz
wget -P input/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/003/SRR1374923/SRR1374923.fastq.gz
wget -P input/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/004/SRR1374924/SRR1374924.fastq.gz
1. 质控
-
使用 FastQC [2] 分析序列质量
“FastQC 旨在提供一种简单的方法来对来自高通量测序管道的原始序列数据进行一些质量检查。它提供了一组模块化的分析,您可以使用它来快速了解您的数据是否存在任何问题。”
处理任何样本之前的第一步是分析数据的质量。fastq 文件中包含质量信息,指的是每个碱基检出的准确度(% 置信度)。FastQC 查看样品序列的不同方面:接头污染、序列重复水平等)
1.1. 安装
-
同时创建新的环境
conda create -n rna-seq -c bioconda fastqc -y
1.2. 运行
fastqc -o results/1_initial_qc/ --noextract input/sample.fastq
1.3. 结果
── results/1_initial_qc/
└── sample_fastqc.html <- 质控结果的 HTML 文件
└── sample_fastqc.zip <- FastQC 报告数据
2. 过滤
-
使用 Trim_Galore [3] 删除低质量序列!
分析数据质量后,下一步是删除不符合质量标准的序列/核苷酸。有大量的质量控制包,但 trim_galore 结合了 Cutadapt[4] 和 FastQC 以删除低质量序列,同时执行质量分析以查看过滤效果。
要选择的 2 个最重要的参数:最小 Phred 分数 (1-30) 和最小测序长度。关于这个参数有不同的看法,您可以查看下面的论文以获取有关使用哪些参数的更多信息。通常是: 20 的 Phred 分数(99% 的置信度)和至少 50-70% 的序列长度。
-
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-0956-2 -
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8 -
http://www.epigenesys.eu/images/stories/protocols/pdf/20150303161357_p67.pdf
2.1. 安装
conda install -c bioconda trim-galore --yes
2.2. 运行
trim_galore --quality 20 --fastqc --length 25 \
--output_dir results/2_trimmed_output/ \
input/sample.fastq
2.3. 结果
── results/2_trimmed_output/
└── sample_trimmed.fq <- 过滤后的测序文件 (.fastq)
└── sample_trimmed.html <- FastQC 质量分析的 HTML 文件
└── sample_trimmed.zip <- FastQC 报告数据
└── sample.fastq.trimming_report.txt <- Cutadapt 修剪报告
3. 去除rRNA
-
使用 [SortMeRNA](http://bioinfo.lifl.fr/RNA/sortmerna/ http://bioinformatics.oxfordjournals.org/content/28/24/3211 "SortMeRNA") 去除 rRNA序列
一旦我们去除了低质量序列和任何接头污染,我们就可以继续执行一个额外的(和可选的)步骤,从样本中去除 rRNA 序列。如果您的样品在文库制备之前未使用 rRNA 去除方案制备,建议运行此步骤以删除任何可能占用大部分比对序列的 rRNA 序列污染。
3.1. 安装
conda install -c bioconda sortmerna --yes
3.2. 创建索引
在我们运行 sortmerna 命令之前,必须首先下载并处理真核、古细菌和细菌 rRNA 数据库。sortmerna_db/ 文件夹将是我们保存运行 SortMeRNA 所需文件的位置。这些数据库只需要创建一次,因此任何未来的 RNAseq 流程中都可以使用这些文件。
# 将 sortmerna 包 下载到 sortmerna_db 文件夹中
wget -P sortmerna_db https://github.com/biocore/sortmerna/archive/2.1b.zip
# 解压文件
unzip sortmerna_db/2.1b.zip -d sortmerna_db
# 将数据库移动到正确的文件夹中
mv sortmerna_db/sortmerna-2.1b/rRNA_databases/ sortmerna_db/
# 删除无用的文件夹
rm sortmerna_db/2.1b.zip
rm -r sortmerna_db/sortmerna-2.1b
# 将所有数据库的位置保存到一个文件夹中
sortmernaREF=sortmerna_db/rRNA_databases/silva-arc-16s-id95.fasta,sortmerna_db/index/silva-arc-16s-id95:\
sortmerna_db/rRNA_databases/silva-arc-23s-id98.fasta,sortmerna_db/index/silva-arc-23s-id98:\
sortmerna_db/rRNA_databases/silva-bac-16s-id90.fasta,sortmerna_db/index/silva-bac-16s-id95:\
sortmerna_db/rRNA_databases/silva-bac-23s-id98.fasta,sortmerna_db/index/silva-bac-23s-id98:\
sortmerna_db/rRNA_databases/silva-euk-18s-id95.fasta,sortmerna_db/index/silva-euk-18s-id95:\
sortmerna_db/rRNA_databases/silva-euk-28s-id98.fasta,sortmerna_db/index/silva-euk-28s-id98
# 创建索引
indexdb_rna --ref $sortmernaREF
3.3. 运行
# 保存 rRNA 数据库的变量
# 将所有数据库的位置保存到一个文件夹中
sortmernaREF=sortmerna_db/rRNA_databases/silva-arc-16s-id95.fasta,sortmerna_db/index/silva-arc-16s-id95:\
sortmerna_db/rRNA_databases/silva-arc-23s-id98.fasta,sortmerna_db/index/silva-arc-23s-id98:\
sortmerna_db/rRNA_databases/silva-bac-16s-id90.fasta,sortmerna_db/index/silva-bac-16s-id95:\
sortmerna_db/rRNA_databases/silva-bac-23s-id98.fasta,sortmerna_db/index/silva-bac-23s-id98:\
sortmerna_db/rRNA_databases/silva-euk-18s-id95.fasta,sortmerna_db/index/silva-euk-18s-id95:\
sortmerna_db/rRNA_databases/silva-euk-28s-id98.fasta,sortmerna_db/index/silva-euk-28s-id98
# 运行 SortMeRNA
sortmerna \
--ref $sortmernaREF \
--reads results/2_trimmed_output/sample_trimmed.fq \
--aligned results/3_rRNA/aligned/sample_aligned.fq \
--other results/3_rRNA/filtered/sample_filtered.fq \
--fastx \
--log \
-a 4 \
-v
# 将日志移动到正确的文件夹中
mv -v results/3_rRNA/aligned//sample_aligned.log results/3_rRNA/logs
3.4. 结果
── results/3_rRNA/
└── aligned/sample_aligned.fq <- rRNA 污染的序列
└── filtered/sample_filtered.fq <- 无任何 rRNA 污染的序列
└── logs/sample_aligned.log <- SortMeRNA 分析的日志
4. 比对
-
使用 STAR-aligner [5] 进行基因组比对
STAR aligner 是一种非常快速有效的拼接比对工具,用于将 RNAseq 数据与基因组进行比对。 STAR aligner 具有发现非规范剪接和嵌合(融合)转录本的能力,但对于我们的用例,我们将使用全长 RNA 序列与基因组进行比对。该工具的输出是一个 .BAM 文件,它表示每个序列已对齐的坐标。 .BAM 文件与 .SAM 文件相同,但它是二进制格式,因此您无法查看内容,这极大地减小了文件的大小。
4.1. 安装
conda install -c bioconda star --yes
4.2. 创建索引
与 SortMeRNA 步骤类似,我们必须首先生成要比对的基因组索引,以便工具可以有效地映射数百万个序列。 star_index 文件夹将是我们运行 STAR 所需文件的位置,并且由于程序的性质,它最多可以占用 30GB 的空间。此步骤只需运行一次,即可用于任何后续的 RNAseq 比对分析。
STAR \
--runMode genomeGenerate \
--genomeDir star_index \
--genomeFastaFiles genome/* \
--sjdbGTFfile annotation/* \
--runThreadN 4
4.3. 运行
# 运行 STAR
STAR \
--genomeDir star_index \
--readFilesIn filtered/sample_filtered.fq \
--runThreadN 4 \
--outSAMtype BAM SortedByCoordinate \
--quantMode GeneCounts
# 将 BAM 文件移动到正确的文件夹中
mv -v results/4_aligned_sequences/sampleAligned.sortedByCoord.out.bam results/4_aligned_sequences/aligned_bam/
# 将日志移动到正确的文件夹中
mv -v results/4_aligned_sequences/${BN}Log.final.out results/4_aligned_sequences/aligned_logs/
mv -v results/4_aligned_sequences/sample*Log.out results/4_aligned_sequences/aligned_logs/
4.4. 结果
── results/4_aligned_sequences/
└── aligned_bam/sampleAligned.sortedByCoord.out.bam <- 排序的 BAM 比对文件
└── aligned_logs/sampleLog.final.out <- STAR 比对率日志
└── aligned_logs/sampleLog.out <- STAR 比对过程中的步骤日志
5. 定量
-
用 featureCounts [6] 总结基因计数
现在有了 .BAM 比对文件,可以继续尝试将这些坐标总结为基因丰度。为此,必须使用 featureCounts 或任何其他读数汇总工具汇总读数,并按具有原始序列丰度的样本生成基因表。然后该表将用于执行统计分析并找到差异表达的基因。
5.1. 安装
conda install -c bioconda subread --yes
5.2. 运行
# 将目录更改为比对的 .BAM 文件夹
cd results/4_aligned_sequences/aligned_bam
# 将文件列表存储为变量
dirlist=$(ls -t ./*.bam | tr '\n' ' ')
echo $dirlist
# 运行 featureCounts
featureCounts \
-a ../../annotation/* \
-o ../../results/5_final_counts/final_counts.txt \
-g 'gene_name' \
-T 4 \
$dirlist
# 将目录更改回主文件夹
cd ../../../
5.3. 结果
── results/5_final_counts/
└── final_counts.txt <- 所有样本的最终基因计数
└── final_counts.txt.summary <- 基因计数总结
6. 质控报告
-
使用 multiQC [7] 生成指控分析报告
在质量过滤、rRNA 去除、STAR 比对和基因定量期间,创建了多个日志文件,其中包含衡量各个步骤质量的指标。我们可以使用汇总工具 MultiQC 来搜索所有相关文件并生成丰富的图表来显示来自不同步骤日志文件的数据,而不是遍历许多不同的日志文件。在确定序列与基因组的比对情况以及确定每个步骤丢失了多少序列时,此步骤非常有用。
6.1. 安装
conda install -c bioconda multiqc --yes
6.2. 运行
# 运行 multiqc 并将结果输出到最终文件夹
multiqc results \
--outdir results/6_multiQC
6.3. 结果
── results/6_multiQC/
└── multiqc_report.html <- 代表每一步的日志结果
└── multiqc_data/ <- multiqc 从各种日志文件中找到的数据文件夹
欢迎Star -> 学习目录
国内链接 -> 学习目录
参考资料
Source: https://github.com/twbattaglia/RNAseq-workflow
[2]FastQC: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
[3]Trim_Galore: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
[4]Cutadapt: http://cutadapt.readthedocs.io/en/stable/guide.html
[5]STAR-aligner: https://www.ncbi.nlm.nih.gov/pubmed/23104886
[6]featureCounts: https://www.ncbi.nlm.nih.gov/pubmed/24227677
[7]multiQC: http://multiqc.info/
本文由 mdnice 多平台发布