单细胞转录组 —— STARsolo 原始数据处理实战
前言
前面我们已经介绍了几种原始数据处理工具,最后再介绍一种多平台兼容的快速定量工具 —— STARsolo。
主要使用的还是 STAR 比对软件,只是增加了更多对单细胞数据的处理,不同平台数据的差异,也只是在参数设置上。
实战
软件可以直接用 conda 进行安装
conda create -n STAR -c bioconda -c conda-forge star seqtk
下载人类参考基因组
wget http://ftp.ensembl.org/pub/release-98/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
wget http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.primary_assembly.annotation.gtf.gz
或者小鼠参考基因组
wget ftp://ftp.ensembl.org/pub/release-98/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz
wget ftp://ftp.ensembl.org/pub/release-98/gtf/mus_musculus/Mus_musculus.GRCm38.98.gtf.gz
解压文件
gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gunzip gencode.v32.primary_assembly.annotation.gtf.gz
gunzip Mus_musculus.GRCm38.dna.primary_assembly.fa.gz
gunzip Mus_musculus.GRCm38.98.gtf.gz
构建索引
构建人类参考基因组索引
STAR \
--runThreadN 20 \
--runMode genomeGenerate \
# 索引保存路径
--genomeDir human \
# fa 文件路径
--genomeFastaFiles Homo_sapiens.GRCh38.dna.primary_assembly.fa \
# gtf文件路径
--sjdbGTFfile gencode.v32.primary_assembly.annotation.gtf
类似地,可以构建小鼠参考基因组索引
STAR \
--runThreadN 20 \
--runMode genomeGenerate \
# 索引保存路径
--genomeDir mmu \
# fa 文件路径
--genomeFastaFiles Mus_musculus.GRCm38.dna.primary_assembly.fa \
# gtf文件路径
--sjdbGTFfile Mus_musculus.GRCm38.98.gtf
scRNA-seq
从 SRA 数据库中下载 PRJNA666791 项目的 10x scRNA-seq 数据进行分析
prefetch --option-file SRR_Acc_List.txt --output-directory sra
解压数据
ls sra/*/*.sra | xargs fastq-dump --split-files --gzip -O raw
10x barcode 文件可以在下载的 cellranger 软件中找到,例如
ls /path/to/cellranger-8.0.0/lib/python/cellranger/barcodes
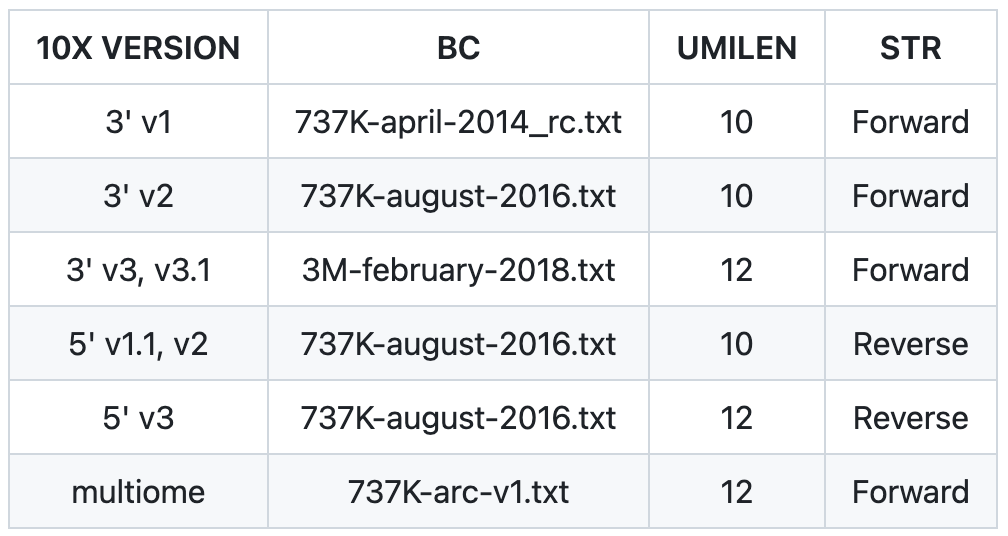
参数配置,需要根据所使用数据的信息进行调整
```bash
# 白名单
BC=737K-august-2016.txt
# barcode 的长度
CBLEN=16
# UMI 的长度
UMILEN=10
# 文库方向
STRAND=Forward # Forward/Reverse/Unstranded
FQDIR="$(pwd)/raw"
TAG="GSM4812353"
R1=""
R2=""
if [[ `find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "_1\.f.*q"` != "" ]]
then
R1=`find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "_1\.f.*q" | sort | tr '\n' ',' | sed "s/,$//g"`
R2=`find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "_2\.f.*q" | sort | tr '\n' ',' | sed "s/,$//g"`
elif [[ `find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "R1\.f.*q"` != "" ]]
then
R1=`find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "R1\.f.*q" | sort | tr '\n' ',' | sed "s/,$//g"`
R2=`find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "R2\.f.*q" | sort | tr '\n' ',' | sed "s/,$//g"`
elif [[ `find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "_R1_.*\.f.*q"` != "" ]]
then
R1=`find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "_R1_.*\.f.*q" | sort | tr '\n' ',' | sed "s/,$//g"`
R2=`find $FQDIR/* | grep -P "\/$TAG[\/\._]" | grep "_R2_.*\.f.*q" | sort | tr '\n' ',' | sed "s/,$//g"`
else
>&2 echo "ERROR: No appropriate fastq files were found! Please check file formatting, and check if you have set the right FQDIR."
exit 1
fi
比对和定量分析,注意 指定文件夹时末尾需要带上路径分隔符(/),例如 output/
STAR --runThreadN 16 \
--genomeDir mmu \
--runDirPerm All_RWX \
--readFilesCommand gunzip -c\
--outSAMtype BAM Unsorted \ # 最好不要在这里排序
--readFilesIn $R2 $R1 \
--soloType CB_UMI_Simple \
--soloCBwhitelist $BC
--soloBarcodeReadLength 0 \
--soloCBlen $CBLEN \
--soloUMIstart $((CBLEN+1)) \
--soloUMIlen $UMILEN \
--soloStrand $STRAND \
--soloUMIdedup 1MM_CR --soloCBmatchWLtype 1MM_multi_Nbase_pseudocounts \
--soloUMIfiltering MultiGeneUMI_CR \
--soloCellFilter EmptyDrops_CR --clipAdapterType CellRanger4 \
--outFilterScoreMin 30 \
--soloFeatures Gene GeneFull Velocyto \
--soloOutFileNames output/ features.tsv barcodes.tsv matrix.mtx \
--soloMultiMappers EM --outReadsUnmapped Fastx
如果出现类似的错误
EXITING because of FATAL ERROR: number of bytes expected from the BAM bin does not agree with the actual size on disk: Expected bin size=2750819958 ; size on disk=310984704 ; bin number=48
不要设置排序选项 "outSAMtype": "BAM SortedByCoordinate",可以跑完之后,再用 samtools sort
结果文件的结果如下,包含 Gene、GeneFull 和 Velocyto 三个文件夹,它们的文件结构是一模一样的。
inDrops
如果跑过前面的流程,可以使用前面下载好的数据。
GSE111672 数据集中包含 6 例原发性胰腺癌组织的单细胞 RNA 测序和空间转录组学,
我们随便选择一个样本 GSM3036909 下载原始数据
prefetch SRR6825055
解压
fastq-dump SRR6825055/SRR6825055.sra --split-files --gzip -O SRR6825055
其中 R1 长度为 35,R2 长度为 51,是 Indrop-seq 的 V2 版,与 V1 相反,所以需要调换文件顺序
barcode 文件可以从 inDrops barcode lists 中下载。
比对和定量
STAR --runThreadN 8 \
--genomeDir human \
--readFilesIn SRR6825055/SRR6825055_1.fastq.gz SRR6825055/SRR6825055_2.fastq.gz \
--readFilesCommand gunzip -c --outReadsUnmapped None \
--outSAMattrRGline ID:indrop SM:indrop \
--runDirPerm All_RWX --outSAMtype BAM Unsorted \
--soloFeatures Gene GeneFull \
--soloCBwhitelist gel_barcode1_list.txt gel_barcode2_list.txt \
--soloAdapterSequence GAGTGATTGCTTGTGACGCCTT \
--soloType CB_UMI_Complex --soloAdapterMismatchesNmax 3 \
--soloCBmatchWLtype 1MM --soloCBposition 0_0_2_-1 3_1_3_8 \
--soloUMIposition 3_9_3_14 --outTmpDir /tmp/tmplm9atu4a/STARtmp \
--outFileNamePrefix solo/indrop/ \
--soloOutFileNames solo/ features.tsv barcodes.tsv matrix.mtx
Drop-seq
数据集中 GSE178612 研究的是 FoxM1 与 Rb 基因在小鼠乳腺癌中的相互作用。
我们选择其中一个样本 GSM5394388,下载其原始数据
prefetch SRR14872449
fastq-dump SRR14872449/SRR14872449.sra --split-files --gzip -O SRR14872449
比对和定量
STAR --runThreadN 8 \
--genomeDir mmu \
--readFilesIn SRR14872449/SRR14872449_2.fastq.gz SRR14872449/SRR14872449_1.fastq.gz \
--readFilesCommand gunzip -c \
--outReadsUnmapped None \
--outSAMattrRGline ID:drop SM:drop
--runDirPerm All_RWX \
--outSAMtype BAM Unsorted \
--soloFeatures Gene GeneFull \
--soloType CB_UMI_Simple --soloCBwhitelist None -\
-soloCBstart 1 --soloCBlen 12 \
--soloUMIstart 13 --soloUMIlen 8 \
--soloBarcodeReadLength 0 --outTmpDir /tmp/tmpkyaiuqwz/STARtmp \
--outFileNamePrefix solo/drop/ \
--soloOutFileNames solo/ features.tsv barcodes.tsv matrix.mtx
SMART-seq2
我们从研究人类皮质球体内星形胶质细胞的成熟数据 GSE99951 中,下载 GSM2665701 样本进行分析
prefetch SRR5676730
解压
fastq-dump SRR5676730/SRR5676730.sra --split-files --gzip -O SRR5676730
可以先对数据进行去接头处理,可以选择常用的 trimmomatic 软件
conda install -c bioconda trimmomatic
去接头
trimmomatic PE -threads 8 \
SRR5676730/SRR5676730_1.fastq.gz SRR5676730/SRR5676730_2.fastq.gz \
SRR5676730/SRR5676730_1.paired.fastq.gz SRR5676730/SRR5676730_1.unpaired.fastq.gz \
SRR5676730/SRR5676730_2.paired.fastq.gz SRR5676730/SRR5676730_2.unpaired.fastq.gz \
ILLUMINACLIP:/path/to/adapters/TruSeq3-PE-2.fa:2:30:10 \
LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
表达定量
STAR --runThreadN 8 \
--genomeDir human \
--readFilesIn SRR5676730/SRR5676730_1.paired.fastq.gz SRR5676730/SRR5676730_2.paired.fastq.gz \
--readFilesCommand gunzip \
-c --outReadsUnmapped None \
--outSAMattrRGline ID:smart2 SM:smart2 \
--runDirPerm All_RWX --outSAMtype BAM Unsorted \
--soloFeatures Gene GeneFull \
--soloType SmartSeq --soloUMIdedup Exact \
--soloStrand Unstranded --limitOutSJcollapsed 10000000 \
--soloCellFilter None \
--outTmpDir /tmp/tmp8n8c3_75/STARtmp \
--outFileNamePrefix solo/smart2/ \
--soloOutFileNames solo/ features.tsv barcodes.tsv matrix.mtx
CEL-Seq2
使用出生后第 4 天小鼠结节神经节胶质细胞单细胞水平的基因数据集 GSE237947。
选择样本 GSM7656793,下载
prefetch SRR25386888
解压数据
fastq-dump SRR25386888/SRR25386888.sra --split-files --gzip -O SRR25386888
定量分析
STAR --runThreadN 8 \
--genomeDir mmu \
--readFilesIn SRR25386888/SRR25386888_2.fastq.gz SRR25386888/SRR25386888_1.fastq.gz \
--readFilesCommand gunzip -c \
--outReadsUnmapped None --runDirPerm All_RWX \
--outSAMattrRGline ID:cel SM:cel \
--soloBarcodeMate 2 \ # 指定 barcode 在哪个序列上
--outSAMtype BAM Unsorted \
--soloFeatures Gene GeneFull \
--soloCBwhitelist None --soloType CB_UMI_Simple \
--clip5pNbases 12 0 \
--soloUMIstart 1 --soloUMIlen 6 \
--soloCBstart 7 --soloCBlen 6 \
--soloUMIdedup Exact \
--outTmpDir /tmp/tmp4fqrnsfm/STARtmp \
--outFileNamePrefix solo/cel/ \
--soloOutFileNames solo/ features.tsv barcodes.tsv matrix.mtx
BD Rhapsody
GSE241462 收集了小鼠胚胎发育几个阶段种约 200~250 个囊胚和 50 个 E5.5 阶段的胚胎。
选择 GSM7729310 样本进行分析
prefetch SRR25734060
解压数据
fastq-dump SRR25734060/SRR25734060.sra --split-files --gzip -O SRR25734060
定量分析
STAR --runThreadN 8 \
--genomeDir mmu \
--readFilesIn SRR25734060/SRR25734060_2.fastq.gz SRR25734060/SRR25734060_1.fastq.gz \
--readFilesCommand gunzip \
-c --outReadsUnmapped None \
--outSAMattrRGline ID:bd_rhapsody SM:bd_rhapsody \
--runDirPerm All_RWX --outSAMtype BAM Unsorted \
--soloFeatures Gene GeneFull \
--soloCBwhitelist bd_rhapsody_barcode1.txt bd_rhapsody_barcode2.txt bd_rhapsody_barcode3.txt \
--soloType CB_UMI_Complex \
--soloUMIlen 8 --soloCBmatchWLtype 1MM \
--soloCBposition 0_0_0_8 0_21_0_29 0_43_0_51 \
--soloUMIposition 0_52_0_59 \
--outTmpDir /tmp/tmpsdqwfkpz/STARtmp \
--outFileNamePrefix solo/bd_rhapsody/ \
--soloOutFileNames solo/ features.tsv barcodes.tsv matrix.mtx