CCA(Canonical Correlation Analysis)和 Harmony 是两种常用于单细胞 RNA 测序(scRNA-seq)数据整合和批次效应校正的方法。
CCA 通过计算两个(或多个)数据集的线性组合,使这些组合之间的相关性最大化,从而找到不同数据集间的共同信号。在单细胞数据整合中,CCA会找到不同批次数据间的“锚点细胞”作为匹配点,这些锚点代表批次间生物学上相似的细胞。主要用于投影多个数据集到一个共享的低维空间中,使不同批次的数据可以被对齐。
Harmony是一种基于潜在变量模型的批次效应校正方法,它通过迭代的方式将数据投影到一个低维空间,并在这个过程中动态调整数据以去除批次效应。Harmony会根据每次迭代后的聚类结果来修正数据的表示,以确保在去除批次效应的同时保留生物学变异。与 CCA 不同的是,Harmony是在降维的潜在空间中进行批次效应校正,而不是直接在原始的高维表达空间中。
整合方式和场景:
CCA 更适合用于识别不同数据集间的共同信号,特别是当数据集的样本类型明显不同(例如不同组织、不同实验)时效果较好。在整合多种数据来源时,CCA的锚点策略可以有效匹配数据集之间的生物学相似性。对于一些大型数据集,CCA 的计算可能会比较耗时,尤其是在包含多个批次和复杂的异质性时。
Harmony 更擅长处理同类型样本(如多个批次或不同平台的同类细胞)的批次效应校正,尤其适合数据集间具有较高相似度的情况,但事实上这个也不是绝对的,目前认为harmony是在任何情况都可以优先尝试的工具。 它在计算效率上通常优于 CCA,因为 Harmony 是在降维后的空间进行调整,数据维度较小,迭代过程也更为快速。Harmony 能够保持更多的生物学变异,适合下游的聚类和差异分析,且易于实现和参数调节。
分析步骤
1、导入之前处理好的数据
rm(list=ls())
library(Seurat)
library(SeuratData)
library(patchwork)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
load('scRNA.Rdata')
2、SeuratV5版本
scRNA[["RNA"]] <- split(scRNA[["RNA"]], f = scRNA$orig.ident)
scRNA
# An object of class Seurat
# 24357 features across 4031 samples within 1 assay
# Active assay: RNA (24357 features, 2000 variable features)
# 13 layers present: counts.sample1, counts.sample2, counts.sample3, counts.sample4, counts.sample5, counts.sample6, scale.data, data.sample1, data.sample2, data.sample3, data.sample4, data.sample5, data.sample6
# 4 dimensional reductions calculated: pca, harmony, umap, tsne
# run standard anlaysis workflow
scRNA <- NormalizeData(scRNA)
scRNA <- FindVariableFeatures(scRNA,selection.method = "vst",
nfeatures = 5000) # features可自定
scRNA <- ScaleData(scRNA)
scRNA <- RunPCA(scRNA)
scRNA <- FindNeighbors(scRNA, dims = 1:20, reduction = "pca")
scRNA <- FindClusters(scRNA, resolution = 2,
cluster.name = "unintegrated_clusters")
scRNA <- RunUMAP(scRNA, dims = 1:30, reduction = "pca", reduction.name = "umap.unintegrated")
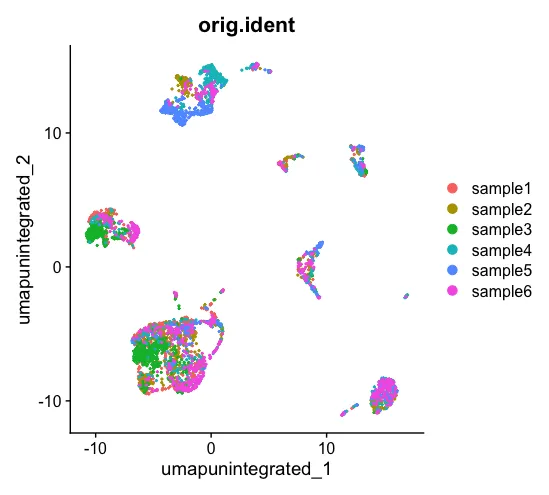
DimPlot(scRNA, reduction = "umap.unintegrated", group.by = c( "orig.ident"))
先不进行CCA整合,看一下UMAP的情况
scRNA <- IntegrateLayers(object = scRNA, method = CCAIntegration,
orig.reduction = "pca", new.reduction = "integrated.cca",
verbose = FALSE)
# re-join layers after integration
scRNA[["RNA"]] <- JoinLayers(scRNA[["RNA"]])
scRNA <- FindNeighbors(scRNA, dims = 1:20, reduction = "integrated.cca")
scRNA <- FindClusters(scRNA, resolution = 2)
scRNA <- RunUMAP(scRNA, dims = 1:30, reduction = "integrated.cca")
DimPlot(scRNA, reduction = "umap", group.by = c("orig.ident"))
CCA整合后,看一下UMAP的情况
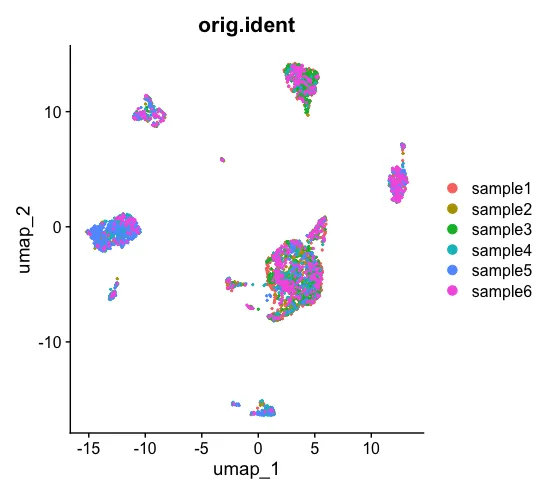
仔细看是会有细微的差别!当然我们在分析之前还是需要提前检查一下是否存在批次问题,然后再做决定是否需要进行整合。
3、SeuratV4版本
# 按照样本进行分割
seurat_list <- SplitObject(scRNA, split.by = "orig.ident")
# 独立对每个数据集的变量特征进行归一化和识别
seurat_list <- lapply(X = seurat_list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst",
nfeatures = 5000) # features可以自行设定
})
# 进行整合
features <- SelectIntegrationFeatures(object.list = seurat_list)
#找到整合的锚点anchors
seurat_anchors <- FindIntegrationAnchors(object.list = seurat_list,
anchor.features = features,
dims = 1:30)
# 利用anchors进行整合
seurat_int <- IntegrateData(anchorset = seurat_anchors, dims = 1:30)
# 降维
#DefaultAssay(seurat_int) <- "integrated"
features = row.names(seurat_int)
seurat_int <- ScaleData(seurat_int,
features = features,
verbose = FALSE
#vars.to.regress = c("percent.mt")
)
seurat_int <- RunPCA(seurat_int, npcs = 30, verbose = FALSE)
seurat_int <- RunUMAP(seurat_int, reduction = "pca", dims = 1:30)
就不展示图片了~
参考资料:
1、Seurat: https://satijalab.org/seurat/articles/integration_introduction.html
2、单细胞天地: https://mp.weixin.qq.com/s/8IJ5NjPzasMDHYFyqwZ22w
3、生信技能树: https://mp.weixin.qq.com/s/i4_kzuAkNZYnB_DfwS-Ppg
4、生信星球: https://mp.weixin.qq.com/s/8ov2wWTecJ7MDyuIKcGkOQ
5、生信菜鸟团:
https://mp.weixin.qq.com/s/QF-nyERJQYSZazUezlsCow
https://mp.weixin.qq.com/s/ZSOnvQ49F6mGz5856gfO1Q
致谢:感谢曾老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -