在生命演进的过程中机体会随着时间的变化而产生不同的变化。从婴幼儿长大为成年人再到老年人的过程中,我们的身体机能经历了从"弱-强-弱"的变化过程(宽泛的说),以年为单位来看,有可能我们在10多岁的时候一年内一下子长高了几十厘米,也有可能在年过百半之后的某一年内突然感觉自己一下子精力大不如前;而以天为单位来看,虽然我们无法从肉眼上看出每个个体在短短24小时有什么显著变化,但事实上我们身体中的某些细胞有可能已经在这二十四小时内过完了它短暂的一生。
这种演进变化从表象来看可以粗暴的归结为“蛋白质的动态变化"(从中心法则来看蛋白质是表征生物体所有特征的最末尾环节),那么再往前推导一下,这种蛋白质的动态变化就是由于不同的基因表达(首先个体的基因组是不变的,但是从纵向来看随着时间的变化基因组的表达可能出现变化,从横向来看不同器官之间的基因组表达也截然不同,以此类推)而导致的。而探究这种演进变化,解码生物体的“动态变化的秘密”就是研究者所关注的重点。
通常情况下,研究者可以在实验的过程中通过多分组情况来构建不同时间点的生物模型,但即使铆足劲了进行分组,人工情况下也绝不可能进行大批量的实验。那么如何来模拟这种情况?拟时序(Pseudotime)/轨迹分析(Trajectory Analysis)就应运而生了。
这种分析方法可以通过测序细胞瞬态情况下的表达模式相似性去构建出细胞的分化/生长/变化轨迹,来模拟细胞动态变化的过程。但这种方法也只能是进行“推断”,甚至很多时候还需要我们具有一定的先验知识去定义变化的起点(或者是选择轨迹)(那这个就挺主观了hhhh),并且要知道导入的数据其实也并不是真正具有时间维度的,只是表示细胞在某个生物过程中中的相对位置(位置由其基因表达模式决定,模拟出了时间维度)。所以笔者有时候觉得这种分析方法只是一种数据的呈现工具hhh,但不可否认的是,这种分析方法的出现也让我们能够进一步得去了解生物体的动态变化,也许在未来随着技术/数据量的提升之后可以做到真正的时序/轨迹分析。
此外我们还需要了解一下它的算法和生物学原理之间的联系,这块在youtube视频资料、曾老师和Hayley的笔记已经详细的整理了(链接见参考资料),笔者就稍作提炼。
1、轨迹分析的关键步骤是降维和轨迹建模,降维是把复杂的数据拆解为不同的关键部分,而这每个关键部分又可以跟生物学上不同的特征相映射在一起,之后再通过定义轨迹的起点和终点来构建出轨迹的形状。
2、降维的方法主要包括线性降维(PCA,ICA)和非线性降维(TSNE,UMAP,DF)。无论是线性还是非线性降维,都是将数据解构,从混杂的信号中分离原始的多个生物信号并跟生物学特征进行映射,而这里面提到的各种方法从算法的角度来说都有其利和弊,只能由使用者自行权衡。
3、轨迹建模的方法也有很多种,目前常用的Monocle2/3分析采用的是反向图嵌入法中的DDRTree。这种方法是先对细胞进行聚类,然后再根据细胞群的平均值的中心点去构建轨迹。同时计算其他细胞到假定轨迹的距离,并将这些细胞分配到在假定轨迹上距离最近的细胞群。同时在这个环节,我们也需要根据先验知识去适当的调整起点位置。
4、单细胞拟时序/轨迹分析把细胞群之间的差异进一步细化到单个细胞的水平并进行展示出来。
分析流程
1、导入
rm(list = ls())
library(Seurat)
library(monocle)
library(dplyr)
load("scRNA.Rdata")
table(Idents(scRNA))
table(scRNA$orig.ident)
head(scRNA@meta.data)
DimPlot(scRNA,label = T)+NoLegend()
2、数据预处理
scRNA$celltype = Idents(scRNA)
# 做拟时序分析通常不是拿全部的细胞,而是拿感兴趣的一部分。用subset提取子集即可。因为要使用差异基因来排序,所以要两类及以上细胞。基于背景知识选择有进化关系的细胞类型。
levels(Idents(scRNA)) #打出来细胞类型供复制
scRNA = subset(scRNA,idents = c("CD8+ T-cells","NK cells"))
table(Idents(scRNA))
##
## CD8+ T-cells NK cells
## 544 92
set.seed(1234)
scRNA = subset(scRNA,downsample = 100)
# 如果只想做一类细胞的拟时序,有两个选择:二次分群再做(和提取两种细胞是一样的道理),或者是选择其他基因用于排序(在后面有)。
# 因为monocle和seurat是两个不同的体系,所以需要将seurat对象转换为monocle可以接受的CellDataSet对象。虽然monocle3已经出来很久了,但大家都不约而同的选择monocle2,大概就是习惯了吧。
3、创建CellDataSet对象
# count矩阵,官方建议用count
ct <- scRNA@assays$RNA$counts
# 基因注释
gene_ann <- data.frame(
gene_short_name = row.names(ct),
row.names = row.names(ct)
)
fd <- new("AnnotatedDataFrame",
data=gene_ann)
# 临床信息
pd <- new("AnnotatedDataFrame",
data=scRNA@meta.data)
#新建CellDataSet对象
sc_cds <- newCellDataSet(
ct,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=1)
sc_cds
4、构建细胞发育轨迹
# 用于估算每个细胞的大小因子,目的是归一化基因表达数据以纠正测序深度的差异
sc_cds <- estimateSizeFactors(sc_cds)
# 估算基因表达的离散度,帮助识别高变异的基因,用于差异基因分析和排序
sc_cds <- estimateDispersions(sc_cds)
# 差异基因分析
dd = "diff.Rdata"
# 完整模型考虑了细胞类型(celltype)和样本来源(orig.ident)
# 简化模型仅考虑样本来源(orig.ident),这样可以检测细胞类型对基因表达的独立影响
if(!file.exists(dd)){
diff_test_res <- differentialGeneTest(sc_cds,
fullModelFormulaStr = " ~ celltype + orig.ident",
reducedModelFormulaStr = " ~ orig.ident",
cores = 4)
save(diff,file = dd)
}
load(dd)
# 筛选用于排序的基因
ordering_genes <- row.names(subset(diff_test_res, qval < 0.01))
#查看基因,筛选适合用于排序的,设置为排序要使用的基因
head(ordering_genes)
length(ordering_genes)
# 设置用于排序的基因
sc_cds <- setOrderingFilter(sc_cds, ordering_genes)
# 画出选择的基因
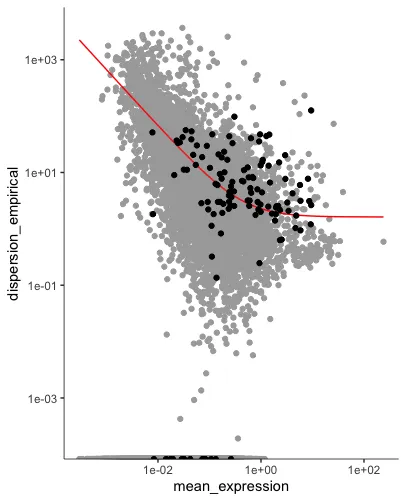
plot_ordering_genes(sc_cds)
# plot_ordering_genes画的图纵坐标是基因表达量的变异性(离散性),横坐标是每个基因在所有细胞种的平均表达量。
# 红线表示Monocle2基于横纵坐标的关系拟合的变异期望值,为用于排序的基因是黑色,其他基因是灰色。
# 对细胞进行降维和去批次,常用于将高维的基因表达数据转换为低维(如 2D)空间,便于可视化和后续分析
sc_cds <- reduceDimension(sc_cds,residualModelFormulaStr = "~orig.ident")
# 根据降维后的数据和排序基因,进行细胞的拟时序排序,推断细胞在轨迹中的位置
sc_cds <- orderCells(sc_cds)
#对比一下单样本和多样本的数据,differentialGeneTest和reduceDimension不一样,要考虑样本。因为我们是从count开始构建CellDataSet对象的,在seurat里面做的批次整合已经无效了,在monocle里面需要重新去除批次效应。
纵坐标是基因表达量的变异性(离散性),横坐标是每个基因在所有细胞种的平均表达量。
红线表示Monocle2基于横纵坐标的关系拟合的变异期望值,为用于排序的基因是黑色,其他基因是灰色。
核心目的是展示用于排序基因的筛选过程。被选为排序的基因(黑色点)在给定表达水平上表现出比预期更高的变异性,这意味着它们能够更好地反映细胞状态的变化,因此适合作为驱动拟时序轨迹推断的关键基因
5、发育轨迹图
library(ggsci)
p1 = plot_cell_trajectory(sc_cds)+ scale_color_nejm()
p2 = plot_cell_trajectory(sc_cds, color_by = 'Pseudotime')
p3 = plot_cell_trajectory(sc_cds, color_by = 'celltype') + scale_color_npg()
library(patchwork)
p2+p1/p3
plot_cell_trajectory(sc_cds, color_by = 'orig.ident')
Pseudotime数值从小到大就是顺序就是推断的发育顺序。图上的点颜色越深,时间越早,颜色越浅,时间越晚。
state是发育的不同阶段,数值越小越靠前。
celltype则可以看到具体的细胞类型在时间轨迹图上的分布。
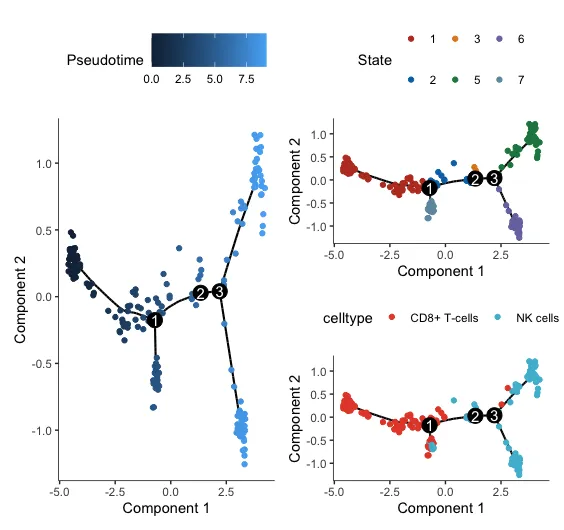
下图是以样本形式的着色轨迹图

6、拟时序热图
gene_to_cluster = diff_test_res %>%
arrange(qval) %>%
head(50) %>%
pull(gene_short_name)
head(gene_to_cluster)
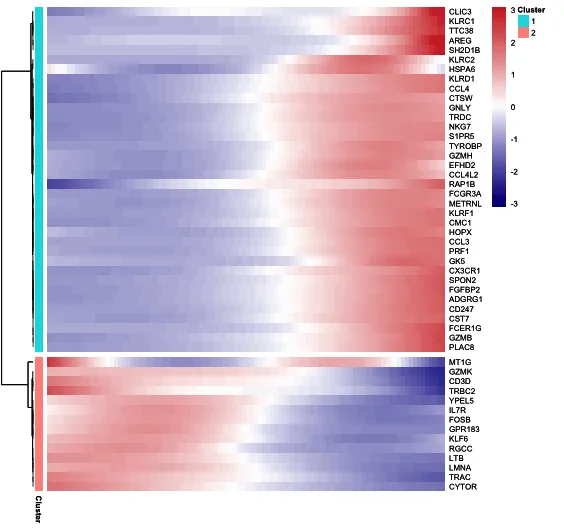
plot_pseudotime_heatmap(sc_cds[gene_to_cluster,],
num_clusters = nlevels(Idents(scRNA)),
show_rownames = TRUE,
cores = 4,return_heatmap = TRUE,
hmcols = colorRampPalette(c("navy", "white", "firebrick3"))(100))
行表示基因:
每一行代表一个基因,这些基因是通过前面差异基因分析筛选出来的显著基因,通常是驱动细胞发育或状态变化的关键基因。
基因名称显示在图的右侧,这些基因在不同的聚类中呈现出不同的表达特征。
列表示细胞顺序:
热图的列按照拟时序(Pseudotime)排序,细胞的排列反映了它们在发育轨迹中的位置,从发育的早期到晚期依次排列。
细胞的顺序反映了细胞在发育过程中逐渐转变的动态变化。
颜色表示基因表达水平:
蓝色:表示基因低表达。白色:表示基因中等表达。红色:表示基因高表达。
颜色的渐变可以帮助我们观察基因表达水平如何随着细胞在发育轨迹中的位置发生变化。
基因的聚类(左侧的树状图):
基因根据表达模式被聚类分组,聚类树(dendrogram)展示了基因之间的相似性。相似表达模式的基因被分在同一个簇中。
例如,图中有明显的两个主要聚类组,每组内的基因在发育过程中表现出类似的表达趋势。
不同的聚类组:
上部聚类组(如 Cluster 1):这些基因在早期(左侧)低表达,随着发育时间的推进(往右),它们逐渐上调(由蓝转红),说明这些基因可能在晚期细胞功能的执行中发挥重要作用。
下部聚类组(如 Cluster 2):这些基因在轨迹的早期高表达(红色),然后逐渐降低(变蓝),表明这些基因可能在早期阶段起重要作用,随后被抑制或关闭。
动态表达模式的生物学意义:
热图中的动态模式反映了细胞在发育过程中的关键转折点。不同簇中的基因可能分别与特定的生物学功能或细胞状态相关,例如细胞增殖、分化、功能成熟等。 通过这些模式,可以推断出哪些基因在特定时间点起调控作用,并识别出可能参与不同发育阶段的信号通路或基因网络。
7、基因轨迹图
# 用感兴趣的基因给轨迹图着色,gs可以换成你想换的基因。
gs = head(gene_to_cluster)
plot_cell_trajectory(sc_cds,markers=gs,
use_color_gradient=T)
横轴与纵轴(Component 1 和 Component 2):
这两个轴是降维后的成分,反映了细胞在拟时序轨迹中的位置。细胞的排列基于其表达特征,展示了不同发育阶段之间的转变。
轨迹与节点(1、2、3):
黑色的轨迹线和标记的节点显示了细胞在拟时序中的轨迹路径。每个节点表示轨迹中的一个关键转折点,通常代表细胞状态的显著变化或分化点。
颜色编码(log10(value + 0.1)):
颜色表示每个基因的表达水平,颜色条从紫色(低表达)到黄色(高表达),并使用对数尺度(log10(value + 0.1))来平滑表达值的可视化。
颜色越接近黄色,表示基因在该细胞中的表达越高;颜色越接近紫色,表示表达越低。
各基因在轨迹中的表达模式:
-
FGFBP2:表达相对较低(多数为紫色),但在轨迹的后期略有上升,表明其在发育晚期可能有一定功能。
-
GNLY:在轨迹的第二和第三节点(2 和 3)之间表达显著升高(由紫色转为黄色),提示其在这一分化或状态转变中起到重要作用。
-
GZMB:表达较为稳定,但在轨迹末端(晚期)略有增加,暗示其在晚期细胞活化或功能执行中可能具有作用。
-
KLRD1:在早期有一定表达,随着轨迹推进呈现下降趋势,暗示其在早期阶段的功能性。
-
NKG7:在轨迹后期(特别是节点 3 附近)表达显著升高,可能与晚期状态的功能关联。
-
TYROBP:与 NKG7 类似,表达在轨迹后期升高,表明其在后期细胞功能中的潜在作用。

8、基因拟时序点图
# 横坐标是按照pseudotime排好顺序的。
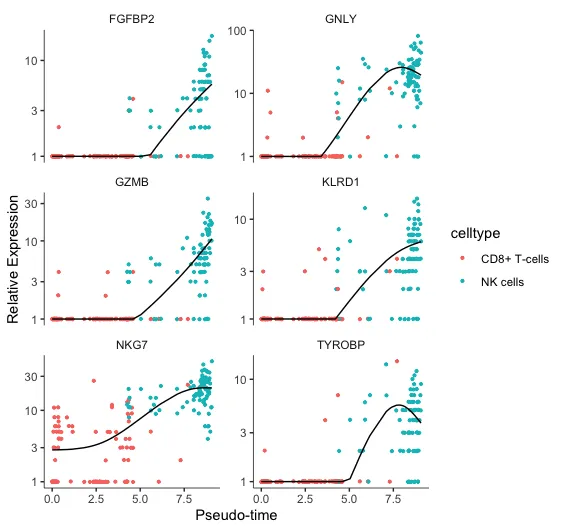
plot_genes_in_pseudotime(sc_cds[gs,],
color_by = "celltype",
nrow= 3,
ncol = NULL )
横轴(Pseudotime):
横轴表示拟时序(Pseudotime),反映了细胞在发育或状态转换中的相对顺序。数值越大,表示细胞处于发育或分化的更晚阶段。
纵轴(Relative Expression):
纵轴表示基因的相对表达水平。采用对数尺度(Log-scale),更好地展示基因表达的动态范围,尤其是低表达和高表达的差异。
不同的基因有不同的表达水平范围,从 1 到 100 或更高,显示了基因在不同阶段的活跃程度。
点的颜色(Celltype):
红色点:表示 CD8+ T-cells。蓝色点:表示 NK cells。
通过点的颜色,可以区分不同细胞类型中基因的表达情况,观察基因在不同细胞类型中的变化模式。
黑色曲线:
黑色曲线表示基因表达随拟时序变化的平滑趋势线。它帮助展示基因在细胞发育过程中的整体表达趋势。
参考资料:
1、生信技能树:
https://mp.weixin.qq.com/s/YHJXm17qPrxuCVsZivHHNw https://mp.weixin.qq.com/s/Q4Vp-bSzLnomggeq-FaMRw https://mp.weixin.qq.com/s/XVG7rXMtq37EW8CsTmwgag https://mp.weixin.qq.com/s/fjZ2rLHB-D64rShOwWxNQQ
2、单细胞天地:
https://mp.weixin.qq.com/s/wzn5Jdf5DR1LrhXecYg7Yw https://mp.weixin.qq.com/s/rgT5VCT0fqvhrTwZOtnaWA
3、Hayley笔记: https://www.jianshu.com/p/83c532234dc0
4、Trajectory inference analysis of scRNA-seq data: https://www.youtube.com/watch?v=XmHDexCtjyw&list=PLjiXAZO27elC_xnk7gVNM85I2IQl5BEJN&index=12
致谢:感谢曾老师、小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -











![[数据结构] 开散列法 闭散列法 模拟实现哈希结构](https://i-blog.csdnimg.cn/direct/2aa0817472f845928d56ebcfd9c88659.png)




