–https://doi.org/10.1038/s41592-024-02201-0
代码来源:https://github.com/bowang-lab/scGPT
学习参考:https://scgpt.readthedocs.io/en/latest/introduction.html
scGPT: Towards Building a Foundation Model for Single-Cell Multi-omics Using Generative AI
留意更多内容,欢迎关注微信公众号:组学之心
研究团队和研究单位
:::: column
::: column-left 60%

:::
::: column-right

:::
::::
scGPT简介
数据:
多种组学:scRNA-seq, scATAC-seq, CITE-seq, Spatial transcriptomics;
33 Million 的单细胞RNA数据 / cross-tissue, human, non-disease;来源于CELLxGENE数据库
数据预处理:先用raw count矩阵,scRNA-seq是RNA分子的read count数目,scATAC-seq则是染色质可及性的peak region。都需要处理成cell-by-gene的矩阵形式。
scGPT的preprocess代码有基本的质控,如filter_gene_by_counts和filter_cell_by_counts
模型主要特点:

- 表达值自适应分箱
- input embedding融合多种信息
- 生成式预训练中独特的注意力掩码机制
- 实现的下游任务丰富
-----数据预处理-质控和分箱-----
Preprocessor自定义函数有一系列参数,如果为False则不执行:
class Preprocessor:
"""
Prepare data into training, valid and test split. Normalize raw expression
values, binning or using other transform into the preset model input format.
"""
def __init__(
self,
use_key: Optional[str] = None,
filter_gene_by_counts: Union[int, bool] = False,
filter_cell_by_counts: Union[int, bool] = False,
normalize_total: Union[float, bool] = 1e4,
result_normed_key: Optional[str] = "X_normed",
log1p: bool = False,
result_log1p_key: str = "X_log1p",
subset_hvg: Union[int, bool] = False,
hvg_use_key: Optional[str] = None,
hvg_flavor: str = "seurat_v3",
binning: Optional[int] = None,
result_binned_key: str = "X_binned",
):
1. 质控过滤
1.1 过滤基因
if self.filter_gene_by_counts:
logger.info("Filtering genes by counts ...")
sc.pp.filter_genes(
adata,
min_counts=self.filter_gene_by_counts
if isinstance(self.filter_gene_by_counts, int)
else None,
)
1.2 过滤细胞
if (
isinstance(self.filter_cell_by_counts, int)
and self.filter_cell_by_counts > 0
):
logger.info("Filtering cells by counts ...")
sc.pp.filter_cells(
adata,
min_counts=self.filter_cell_by_counts
if isinstance(self.filter_cell_by_counts, int)
else None,
)
1.3 normalize
if self.normalize_total:
logger.info("Normalizing total counts ...")
normed_ = sc.pp.normalize_total(
adata,
target_sum=self.normalize_total
if isinstance(self.normalize_total, float)
else None,
layer=key_to_process,
inplace=False,
)["X"]
key_to_process = self.result_normed_key or key_to_process
_set_obs_rep(adata, normed_, layer=key_to_process)
1.4 log1p
if self.log1p:
logger.info("Log1p transforming ...")
if is_logged:
logger.warning(
"The input data seems to be already log1p transformed. "
"Set `log1p=False` to avoid double log1p transform."
)
if self.result_log1p_key:
_set_obs_rep(
adata,
_get_obs_rep(adata, layer=key_to_process),
layer=self.result_log1p_key,
)
key_to_process = self.result_log1p_key
sc.pp.log1p(adata, layer=key_to_process)
这里key_to_process = self.result_log1p_key代码修改了原本key_to_process的值(一开始参数选定是adata.X,存储raw data的位置,经过log1p步骤之后变成了归一化且log1p之后的矩阵)。所以后续的分箱是对归一化log1p后的矩阵来进行。
1.5 hvg挑选
if self.subset_hvg:
logger.info("Subsetting highly variable genes ...")
if batch_key is None:
logger.warning(
"No batch_key is provided, will use all cells for HVG selection."
)
sc.pp.highly_variable_genes(
adata,
layer=self.hvg_use_key,
n_top_genes=self.subset_hvg
if isinstance(self.subset_hvg, int)
else None,
batch_key=batch_key,
flavor=self.hvg_flavor,
subset=True,
)
下一步就是进行表达值分箱了,在下面embedding中一起提到。
–模型框架之Input Embeddings–
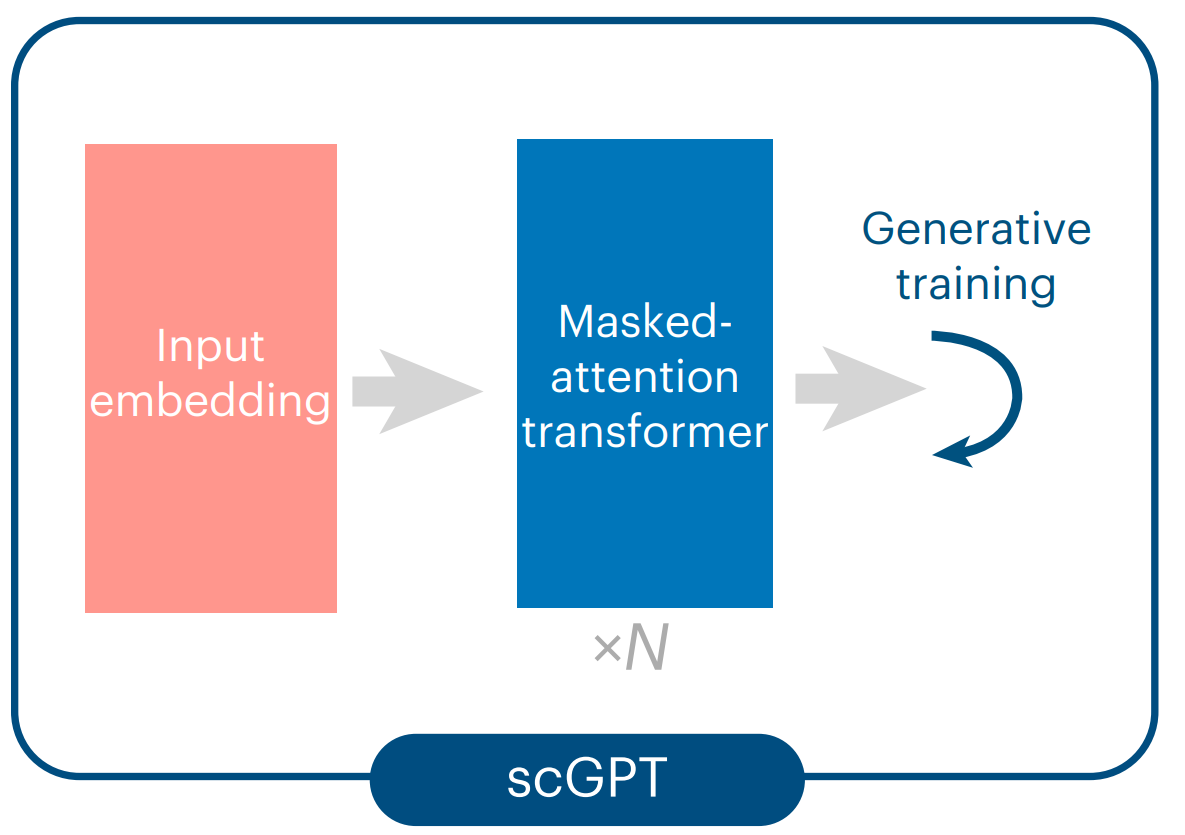
1. Input Embeddings
Input Embeddings主要由三种token组成:
- Gene/peak token
- 表达值token
- 条件token。
输入的矩阵 X ∈ R N × G X∈ ℝ^{N×G} X∈RN×G 需要处理成cell-by-gene矩阵形式,其中 N N N 是细胞数量, G G G 是最初基因数量。

1.1 Gene tokens
把基因symbol名字作为tokens,并为每个基因 g j g_j gj 分配一个id( g j g_j gj)作为词汇表。词汇表还有特殊标记符,如 [cls]。所以每个细胞 i i i 的输入基因tokens由以下公式来表示(M是定义的最大输入长度,就是基因数量的最大值) t g ( i ) ∈ N M t^{(i)}_g ∈ ℕ^M tg(i)∈NM:

之后把每个细胞的gene token通过GeneEncoder嵌入后,转换成 N M × D ℕ^{M×D} NM×D 维度:
class GeneEncoder(nn.Module):
def __init__(
self,
num_embeddings: int,
embedding_dim: int,
padding_idx: Optional[int] = None,
):
super().__init__()
self.embedding = nn.Embedding(
num_embeddings, embedding_dim, padding_idx=padding_idx
)
self.enc_norm = nn.LayerNorm(embedding_dim)
def forward(self, x: Tensor) -> Tensor:
x = self.embedding(x) # (batch, seq_len, embsize)
x = self.enc_norm(x)
return x
1.2 表达值 tokens
用自适应值分箱方法,将每个细胞中的基因表达值进行离散化处理,并转换为相对值:
针对每个细胞中基因表达值非零部分的分布将它们划分为 B 个连续的 bin 区间 [ b k , b k + 1 ] [b_k, b_k+1] [bk,bk+1],其中 k ∈ {1,2,…,B},这个B值是自定义的参数(代码在下面第二部分提到)。
大致意思是把非零基因表达值从小到大排列,按照bin的区间计算分位点,再计算分位点在值队伍中的位置,确定上界和下界,再把基因放进去。
分到相同区间的基因不管之前的表达值具体是多少,分完后都一样。这个分箱过程是针对每个细胞独立完成的,因此 b k b_k bk 会因细胞而异。细胞 i 的分箱基因值 x j ( i ) x^{(i)}_j xj(i) 定义为:
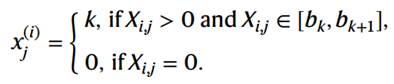
这里和上一章scBERT提到的bag-of-words思想一致,只是分箱的方法不一样。类似拔叔教我们怎么做人,对每样材料依据ta自己的特点进行解肢,然后定制一个个箱子,把材料放进去保存,使得材料能够刚好均匀分布在全部箱子中。
对于微调任务,在分箱步骤之前做了 log1p 转换和 HVG 选择。为了简化表示法,就在分箱之前使用 X i , j X_{i,j} Xi,j 来表示raw count矩阵或预处理数据矩阵。因此,细胞 i 的分箱基因表达值的最终输入向量表示为:
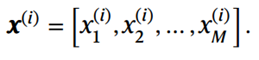
表达值就有3种value encoder,分别对应表达值是category, continuous, scaling的情况。模型默认使用continuous模式,对应着单细胞表达矩阵。作用也是把原本 1 × M 1×M 1×M 维的基因表达值的最终输入向量变成 M × D M×D M×D 维的embedding
CategoryValueEncoder(和GeneEncoder一样):
class CategoryValueEncoder(nn.Module):
def __init__(
self,
num_embeddings: int,
embedding_dim: int,
padding_idx: Optional[int] = None,
):
super().__init__()
self.embedding = nn.Embedding(
num_embeddings, embedding_dim, padding_idx=padding_idx
)
self.enc_norm = nn.LayerNorm(embedding_dim)
def forward(self, x: Tensor) -> Tensor:
x = x.long()
x = self.embedding(x) # (batch, seq_len, embsize)
x = self.enc_norm(x)
return x
ContinuousValueEncoder(2层神经网络):
class ContinuousValueEncoder(nn.Module):
"""
Encode real number values to a vector using neural nets projection.
"""
def __init__(self, d_model: int, dropout: float = 0.1, max_value: int = 512):
super().__init__()
self.dropout = nn.Dropout(p=dropout)
self.linear1 = nn.Linear(1, d_model)
self.activation = nn.ReLU()
self.linear2 = nn.Linear(d_model, d_model)
self.norm = nn.LayerNorm(d_model)
self.max_value = max_value
def forward(self, x: Tensor) -> Tensor:
"""
Args:
x: Tensor, shape [batch_size, seq_len]
"""
# TODO: test using actual embedding layer if input is categorical
# expand last dimension
x = x.unsqueeze(-1)
# clip x to [-inf, max_value]
x = torch.clamp(x, max=self.max_value)
x = self.activation(self.linear1(x))
x = self.linear2(x)
x = self.norm(x)
return self.dropout(x)
Scaling(src先通过GeneEncoder然后存储在cur_gene_token_embs中,values通过valueEncoder后,根据input_emb_style来确定src和values之间的运算关系,一般是相加):
def _encode(
self,
src: Tensor,
values: Tensor,
src_key_padding_mask: Tensor,
batch_labels: Optional[Tensor] = None, # (batch,)
) -> Tensor:
self._check_batch_labels(batch_labels)
src = self.encoder(src) # (batch, seq_len, embsize)
self.cur_gene_token_embs = src
values = self.value_encoder(values) # (batch, seq_len, embsize)
if self.input_emb_style == "scaling":
values = values.unsqueeze(2)
total_embs = src * values
else:
total_embs = src + values
if getattr(self, "dsbn", None) is not None:
batch_label = int(batch_labels[0].item())
total_embs = self.dsbn(total_embs.permute(0, 2, 1), batch_label).permute(
0, 2, 1
) # the batch norm always works on dim 1
elif getattr(self, "bn", None) is not None:
total_embs = self.bn(total_embs.permute(0, 2, 1)).permute(0, 2, 1)
output = self.transformer_encoder(
total_embs, src_key_padding_mask=src_key_padding_mask
)
return output # (batch, seq_len, embsize)
1.3 条件 tokens
条件tokens包含与单个基因相关的各种meta信息,例如扰动实验改变(由扰动标记表示)。为了表示位置条件tokens,使用与输入基因相同维度的输入向量:

类似的有一个BatchLabelEncoder(和GeneEncoder一样):
class BatchLabelEncoder(nn.Module):
def __init__(
self,
num_embeddings: int,
embedding_dim: int,
padding_idx: Optional[int] = None,
):
super().__init__()
self.embedding = nn.Embedding(
num_embeddings, embedding_dim, padding_idx=padding_idx
)
self.enc_norm = nn.LayerNorm(embedding_dim)
def forward(self, x: Tensor) -> Tensor:
x = self.embedding(x) # (batch, embsize)
x = self.enc_norm(x)
return x
1.4 Embedding 层
e m b g emb_g embg 和 e m b c emb_c embc 分别表示 基因token 和 条件token。使用全连接层将低维离散的分箱基因表达值引入非线性表示,学习基因表达模式(表示为 e m b x emb_x embx),以增强表现力(就是上面提到的ContinuousValueEncoder)。这种选择可以对基因表达值的序数关系进行建模:
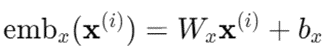
细胞 i i i 的 final embeddings — h ( i ) ∈ R M × D h^{(i)}∈ ℝ^{M×D} h(i)∈RM×D定义为三种 e m b emb emb 的逐元素相加:
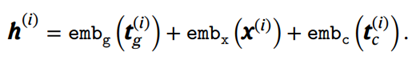
2. 自适应分箱
2.1 执行分箱与变量初始化
if self.binning:
logger.info("Binning data ...")
if not isinstance(self.binning, int):
raise ValueError(
"Binning arg must be an integer, but got {}.".format(self.binning)
)
n_bins = self.binning # NOTE: the first bin is always a spectial for zero
binned_rows = []
bin_edges = []
layer_data = _get_obs_rep(adata, layer=key_to_process)
layer_data = layer_data.A if issparse(layer_data) else layer_data
if layer_data.min() < 0:
raise ValueError(
f"Assuming non-negative data, but got min value {layer_data.min()}."
)
- binned_rows 用于存储所有行的分箱结果。
- bin_edges 用于存储每个行的分箱边界。
- layer_data 获取指定层的数据,并处理稀疏矩阵,检查数据是否包含负值。
2.2 逐行处理数据
for row in layer_data:
if row.max() == 0:
logger.warning(
"The input data contains all zero rows. Please make sure "
"this is expected. You can use the `filter_cell_by_counts` "
"arg to filter out all zero rows."
)
binned_rows.append(np.zeros_like(row, dtype=np.int64))
bin_edges.append(np.array([0] * n_bins))
continue
- 代码遍历 layer_data 中的每一行(一个细胞的表达数据)。
- 如果细胞基因表达全为零,则记录一个警告,并将这行标记为零箱,存储在 binned_rows 中,同时将所有边界设为零,存储在bin_edges中。所以预训练没有去掉无基因表达的细胞。
2.3 计算分箱边界和离散化数据
non_zero_ids = row.nonzero()
non_zero_row = row[non_zero_ids]
bins = np.quantile(non_zero_row, np.linspace(0, 1, n_bins - 1))
# bins = np.sort(np.unique(bins))
# NOTE: comment this line for now, since this will make the each category
# has different relative meaning across datasets
non_zero_digits = _digitize(non_zero_row, bins)
assert non_zero_digits.min() >= 1
assert non_zero_digits.max() <= n_bins - 1
binned_row = np.zeros_like(row, dtype=np.int64)
binned_row[non_zero_ids] = non_zero_digits
binned_rows.append(binned_row)
bin_edges.append(np.concatenate([[0], bins]))
-
首先提取出非零的基因及其索引。
-
先通过np.linspace(0, 1, n_bins - 1) 生成 n_bins-1 个均匀间隔的分箱边界,然后np.quantile计算这些非零基因表达值的分位数,再用自定义的_digitize函数计算分箱区间,将数据分配到一组给定的区间中:
def _digitize(x: np.ndarray, bins: np.ndarray, side="both") -> np.ndarray:
"""
Digitize the data into bins. This method spreads data uniformly when bins
have same values.
Args:
x (:class:`np.ndarray`):
The data to digitize.
bins (:class:`np.ndarray`):
The bins to use for digitization, in increasing order.
side (:class:`str`, optional):
The side to use for digitization. If "one", the left side is used. If
"both", the left and right side are used. Default to "one".
Returns:
:class:`np.ndarray`:
The digitized data.
"""
assert x.ndim == 1 and bins.ndim == 1
left_digits = np.digitize(x, bins)
if side == "one":
return left_digits
right_difits = np.digitize(x, bins, right=True)
rands = np.random.rand(len(x)) # uniform random numbers
digits = rands * (right_difits - left_digits) + left_digits
digits = np.ceil(digits).astype(np.int64)
return digits
- 将非零数据转换为对应的分箱(1 到 n_bins-1),零值保留为零。
- 将结果存储到 binned_row(分箱后的数据)和 bin_edges(分箱边界)中,并添加到对应的列表中。
提一嘴:最后细胞i的输入embedding是:
3. 通过 scGPT transformer 对细胞和基因表达建模
用自注意力机制transformer对 h ( i ) h^{(i)} h(i) 进行encode得到 h n ( i ) ∈ R M , D h^{(i)}_n∈ ℝ^{M,D} hn(i)∈RM,D:

将scGPT-transformer得到的 h n ( i ) ∈ R M , D h^{(i)}_n∈ ℝ^{M,D} hn(i)∈RM,D 直接用于基因水平的微调任务:基因表达预测(GEP)和扰动表达预测任务(perturb-GEP)。
而对于细胞水平任务,需要先将 h n ( i ) h^{(i)}_n hn(i) 整合到细胞embedding向量(3.1细胞表示)中,任务有细胞类型注释,即细胞嵌入通过分类器后预测细胞类型标签。
输入维度 M 可以达到数万个基因,超过了 NLG 中常用的常规transformer的输入长度。使用 FlashAttention的加速自我注意力机制实现,增强模型容量,并支持对大型输入维度的有效处理。其它任何高效的transformer也可以用,例如Linformer和内核自注意力(KSA)。
3.1 细胞表示
细胞i表示 h c ( i ) ∈ R D h^{(i)}_c∈ ℝ^D hc(i)∈RD 是在基因水平表示 h n ( i ) h^{(i)}_n hn(i)中得到的。各种池化操作,如逐元素均值池化或加权池化,都可以在这种情况下使用。[cls] token 是在 Transformer 模型中常用的一种特殊标记符,用来表示整个输入序列的整体信息。
在基因表达模型中,将 [cls] token 附加到input token的开头。这个 [cls] token 的嵌入向量最终将包含整个细胞的综合信息。经过 Transformer 层的处理后,细胞嵌入 h c ( i ) h^{(i)}_c hc(i) 可以由堆叠的 h n ( i ) h^{(i)}_n hn(i) [cls]中的相应行提取出来。
[cls] 操作其实就是检索 [cls] token位置索引处的行并提取出来。
def _get_cell_emb_from_layer(
self, layer_output: Tensor, weights: Tensor = None
) -> Tensor:
"""
Args:
layer_output(:obj:`Tensor`): shape (batch, seq_len, embsize)
weights(:obj:`Tensor`): shape (batch, seq_len), optional and only used
when :attr:`self.cell_emb_style` is "w-pool".
Returns:
:obj:`Tensor`: shape (batch, embsize)
"""
if self.cell_emb_style == "cls":
cell_emb = layer_output[:, 0, :] # (batch, embsize)
elif self.cell_emb_style == "avg-pool":
cell_emb = torch.mean(layer_output, dim=1)
elif self.cell_emb_style == "w-pool":
if weights is None:
raise ValueError("weights is required when cell_emb_style is w-pool")
if weights.dim() != 2:
raise ValueError("weights should be 2D")
cell_emb = torch.sum(layer_output * weights.unsqueeze(2), dim=1)
cell_emb = F.normalize(cell_emb, p=2, dim=1) # (batch, embsize)
return cell_emb
3.2 批次和模态的表示(类似Condition tokens)
使用标准嵌入层以类似的方式实现(比如上面提到的GeneEncoder和BatchLabelEncoder)。这一步主要是想缓解批次效应和整合多模态数据。
模态(modality)token t m ( i ) t^{(i)}_m tm(i) 与各个输入特征 g j g_j gj 相关联(例如,指示它是基因、region还是蛋白质)。批次(batch)token最初位于细胞水平,但也可以传播到单个细胞的所有特征。相同的批次token t b ( i ) t^{(i)}_b tb(i)可以重复,直到单个细胞 i i i 的输入特征长度达到 M M M:

这些批次和模态token的嵌入不用作transformer块的输入。它们在进入特定的微调目标之前,在特征或细胞级别与transformer输出连接起来。
这是为了防止transformer放大相同模态特征内的注意力,而低估不同模态的特征。此外,了解模态和/或批次身份有助于在下游微调目标中进行基因表达建模。当模型学习预测以模态和/或批次身份为条件的表达值时,这种偏差会从基因和细胞表示本身中隐式消除。这是一种促进批次校正的技术。
在单细胞多组学整合任务中,将transformer输出与batch和modality embeddings的和连接起来( e m b b emb_b embb 和 e m b m emb_m embm 先逐元素相加,再和 h n ( i ) h^{(i)}_n hn(i)拼接起来)得到 h n ′ ( i ) h^{'(i)}_n hn′(i),用作表达建模的下游微调目标的输入:

而只有scRNA-seq一种模态,但是有不同的批次,面对scRNA-seq整合任务的时候,只用 h c ( i ) h^{(i)}_c hc(i) 和 e m b b emb_b embb 拼接起来得到 h c ′ ( i ) h^{'(i)}_c hc′(i):

上面的式子中, t b ( i ) t^{(i)}_b tb(i) 表示细胞 i i i 的批次同一性, h c ( i ) h^{(i)}_c hc(i) 是微调目标中的原始细胞表示。请注意,修改后的版本 h c ′ ( i ) h^{'(i)}_c hc′(i) 仅与表达式建模目标相关,不适用于基于分类的目标。
–模型框架之Masked-attention–
1.一些背景
自注意力机制被广泛用于捕捉词汇之间的共现模式。在自然语言处理中,主要通过以下两种方式实现:
-
①掩码token预测,主要用于如BERT和RoBERTa等Transformer编码器模型中。在这种方法中,输入序列中的一些token会被随机掩码,然后模型会在输出中预测这些标记。
-
②自回归生成,通过因果Transformer解码器模型(如OpenAI GPT)进行顺序预测。
在OpenAI GPT-3和GPT-4中使用的生成预训练采用了统一框架,在该框架中,模型预测最有可能的下一个token,该token来自于已知输入token的“prompts”。这个框架提供了高度的灵活性,可以用于各种自然语言生成应用,并展示了诸如上下文感知等能力。scGPT研究认为,生成训练也可以在单细胞模型中以类似的方式发挥作用。
具体来说,scGPT研究对以下两个任务感兴趣:
-
1.基于已知基因表达生成未知的基因表达值,即“基因prompts”的生成;
-
2.在给定输入细胞类型条件下生成全基因组表达值,即“细胞prompts”的生成。
尽管token和prompts的概念相似,但由于数据的非顺序性,基因读取与自然语言的建模有本质区别。与句子中的词语不同,细胞中基因的顺序是可以互换的,因此不存在“下一个基因”来预测的概念。这使得将GPT模型的因果掩码公式不能直接应用于单细胞数据。因此,scGPT研究开发了一种专门的注意力掩码机制,定义了基于注意力分数的预测顺序。
2.注意力掩码
这部分代码较长,可以在github上的model.py中FlashTransformerEncoderLayer和FlashTransformerEncoderLayer中学习~
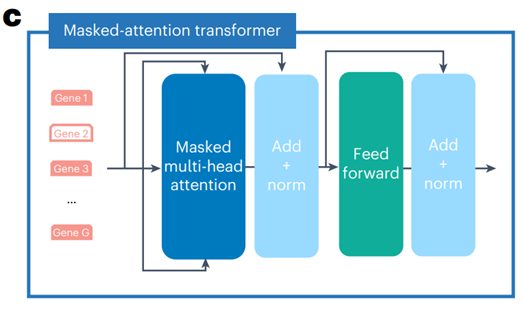
注意力掩码可以应用于 transformer 块中的自注意力矩阵,如对于 M M M 个基因的input token:

第(l + 1)个 transformer块对其输入 M M M 个token的 h l ( i ) ∈ R M , D h^{(i)}_l∈ ℝ^{M,D} hl(i)∈RM,D 应用多头自我注意, h l ( i ) h^{(i)}_l hl(i)是细胞的final embedding h ( i ) h^{(i)} h(i) 经过transformer块的输出:

具体来说,每个self-attention操作的计算方式如下:
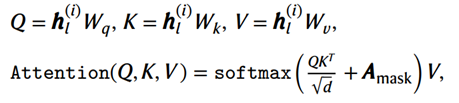
其中 Q , K , V ∈ R M × D Q, K, V ∈ ℝ^{M×D} Q,K,V∈RM×D , W q , W k , W v ∈ R M × d Wq, Wk, Wv ∈ ℝ^{M×d} Wq,Wk,Wv∈RM×d, A m a s k ∈ [ 0 , − i n f ] M × M A_{mask} ∈ [0,− inf]^{M×M} Amask∈[0,−inf]M×M。 A m a s k A_{mask} Amask 中 Q Q Q 是行, K K K 是列(需要符合上面公式中 Q K T QK^T QKT 的维度)
注意力掩码 A m a s k A_{mask} Amask 通过“灭活” Q Q Q 和 K K K 之间的原始注意力权重来修改自我注意力的范围:将 − i n f − inf −inf 添加到矩阵中的位置 ( i , j ) (i,j) (i,j) 会使 softmax 之后的注意力权重无效,从而禁止第 i 个查询和第 j 个键之间的注意力。另一方面,添加 0 意味着注意力权重保持不变。
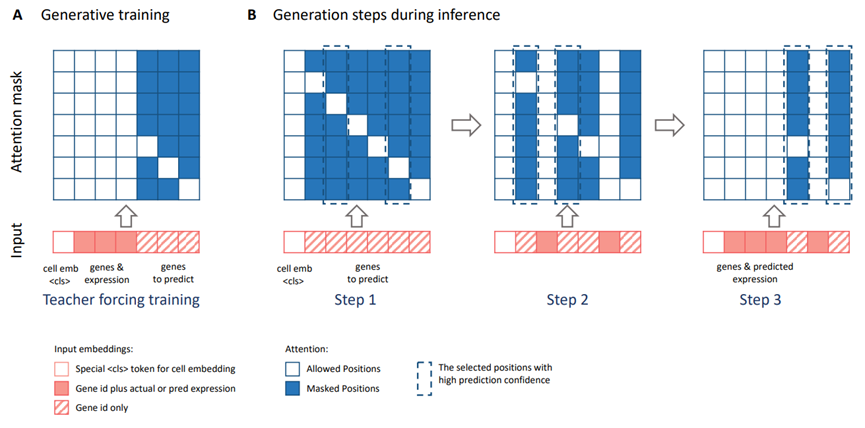
A m a s k A_{mask} Amask如上图A所示,scGPT注意力掩码的原则是仅允许在 “已知基因” 的 embedding 和基因 Q Q Q 之间进行注意力计算,这是通过 A m a s k A_{mask} Amask 的 a i , j a_{i,j} ai,j来实现的,对未知基因(需要预测的基因)的 Q K T QK^T QKT注意力矩阵进行掩码(蓝色),但是从左上到右下的对角线上的区域不掩码:

每个输入嵌入向量 h l ( i ) h^{(i)}_l hl(i)中的token是保留的[cls]标记,用于细胞嵌入;具有已知基因symbol token嵌入和基因表达值嵌入;需要预测表达值的未知基因。
3.掩码后的计算
因果掩码设计:在每次生成迭代中,scGPT预测一组新基因的表达值,而这些基因在下次迭代中会成为“已知基因”,用于下一步的注意力计算,即通过逐步预测非顺序的单细胞数据来实现下一个token预测。在训练过程中随机选择一部分基因作为未知基因,因此它们的表达值在输入中被忽略。
scGPT模型通过堆叠的Transformer块与上述的掩码注意力图预测这些未知基因的表达。在进行细胞prompts生成时,scGPT根据特定的细胞类型生成全基因组表达值。训练好的细胞嵌入被输入,放置在代表细胞类型条件的第一个位置。整个基因表达生成过程分为 K K K 次迭代步骤(即上图b中K=3步)。
例如,在一次迭代中 𝑖∈[1,2,…,𝐾],注意力掩码机制允许与前 0≤𝑖−1次迭代中预测的所有基因进行注意力计算。在每次迭代中,scGPT选择未知集合中预测置信度最高的前 1/K 的基因,以作为下次迭代中的已知基因。
直观地,这种工作流通过自回归方式简化了基因表达的生成,其中预测置信度最高的基因表达值首先生成,并用于帮助后续轮次的生成。
基因prompts生成以类似的方式进行。不同之处在于它以一组具有观察到的表达值的已知基因开始,而不是细胞嵌入。scGPT注意力掩码统一了已知基因编码过程和未知基因生成过程,也成为首个在非顺序数据中执行自回归生成的Transformer架构之一。
4.预训练的学习目标
预训练中的学习目标是通过估计未知基因的表达值来优化模型。这通过使用多层感知器(MLP)来实现,MLP的任务是根据模型的输入预测基因表达值,并通过计算预测值与真实值之间的均方误差来衡量模型的性能,并计算均方误差损失 𝐿:
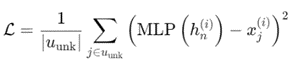
其中,𝑢_unk 表示未知基因的输出位置集合,𝑥_𝑗^{(𝑖)}是要预测的实际基因表达值。∣⋅∣操作获取集合中的元素数量。
在训练过程中,基因prompts和细胞prompts生成这两种模式连续进行。对于给定细胞的输入基因标记,一部分基因被选为“未知”基因,它们的表达值被忽略。
-
在基因prompts步骤中,模型的输入包括[cls]token嵌入、已知基因嵌入和未知基因嵌入。损失使用模型的输出计算。
-
在细胞prompts步骤中,使用先前步骤输出的细胞嵌入(即 ℎ_𝑐^{(𝑖)}在细胞表示中)替换[cls]位置的嵌入,其他计算保持相同。
最后,两种步骤的损失值相加,并用于计算梯度以优化模型参数。整个过程是连续进行的,通过这种方式,模型不仅可以学习预测未知基因的表达值,还能根据细胞的上下文信息进行更精准的表达预测。
–微调目标–
1.基因表达预测(GEP)
这个微调目标与预训练中的目标类似,但应用于掩码位置。具体来说,对于每个输入细胞,一部分基因标记及其对应的表达值 𝑥^{(𝑖)} 会被随机掩码。scGPT通过优化准确预测这些掩码位置的表达值来达到微调目的。这一微调目标有助于模型有效编码数据集中基因间的共表达关系。
该目标通过最小化掩码位置的均方误差来实现,表示为 𝑀_{mask}。GEP的工作原理如下:这里x̃ ^{(i)} ∈ℕ^M,表示细胞 i的表达估计的行。如果有批次或者模态条件信息,用h{'(i)}_n来代替h{(i)}_n
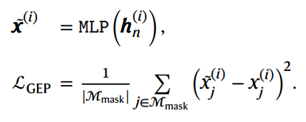
GEP是一种通用的自监督微调目标,旨在预测基因表达值。在某些下游任务中,如扰动预测,模型需要预测基因扰动后的表达值,而不是原始值。研究将这种变体称为扰动GEP。保留上面方程中的MLP估算器,但使用扰动后基因表达作为目标值 𝑥^{(𝑖)}_j。在扰动GEP中,模型的任务是预测所有输入基因的扰动后表达。
2.用于细胞建模的基因表达预测
这个微调目标与GEP类似,但基于细胞表示h^{(i)}_c来预测基因表达值,从而促进细胞表示学习。对于每个细胞 𝑖中的基因 𝑗,我们创建一个查询向量 𝑞_𝑗,并使用 𝑞_𝑗和细胞表示的 h^{(i)}_c 参数化内积作为预测的表达值
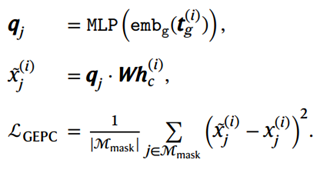
3.弹性细胞相似性
这个微调目标通过使用相似性学习损失来增强细胞表示:
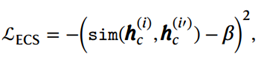
其中 sim代表余弦相似度函数,𝑖和 𝑖^′指代小批量中的两个细胞。此外,𝛽表示一个预定阈值,而 ECS是弹性细胞相似性。这个方法背后的思想是通过提升相似性高于 𝛽的配对,使其更加相似;相反,不相似的配对则被鼓励进一步分离。
4.通过反向传播来实现领域自适应
细胞表示学习受限于批次效应的存在,这些效应是由于测序技术引入的非生物学批次差异所导致的。为了缓解这个问题,scGPT使用了一个独立的MLP分类器来预测与每个输入细胞 ℎ_𝑐^{(𝑖)}相关的测序批次,并通过在模型内反转梯度来修改反向传播过程。
5.细胞类型分类
这个微调目标旨在利用学习到的细胞表示来注释单个细胞。scGPT使用一个独立的MLP分类器根据细胞表示 ℎ_𝑐^{(𝑖)} 预测细胞类型。这个微调目标通过预测的细胞类型概率与真实标签之间的交叉熵损失来优化。
–实操应用–
scGPT需要Python >= 3.7.13 and R >=3.6.1,而且scGPT已经上传在PyPI中,而且在HuggingFace中提供了运行预训练工作流程的初步支持
https://github.com/bowang-lab/scGPT/tree/integrate-huggingface-model
pip install scgpt "flash-attn<1.0.5"
pip install wandb
git clone this-repo-url
cd scGPT
poetry install
预训练模型的列表,可以获取训练好的权重文件:
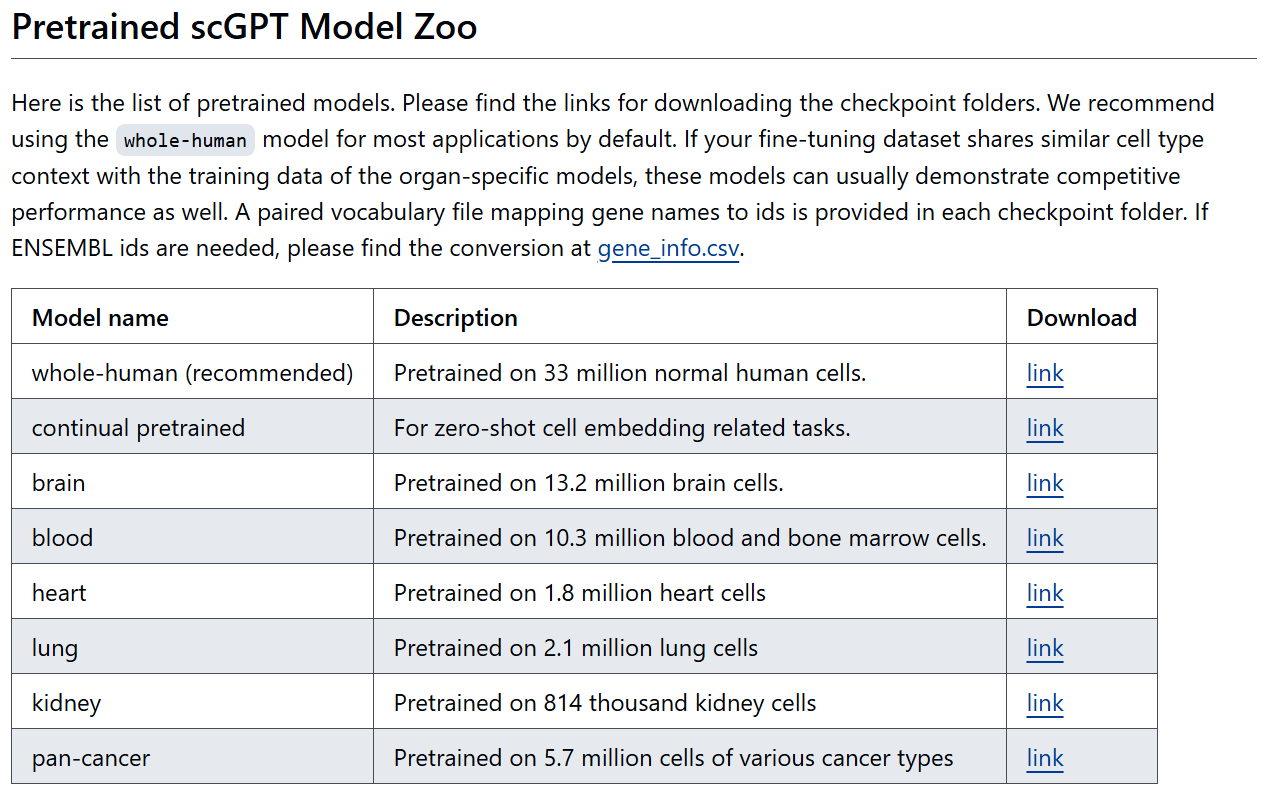
作者还提供了微调模型的练习代码文件
https://github.com/bowang-lab/scGPT/blob/main/examples/finetune_integration.py
https://scgpt.readthedocs.io/en/latest/introduction.html
–下游任务应用–
1.细胞类型注释

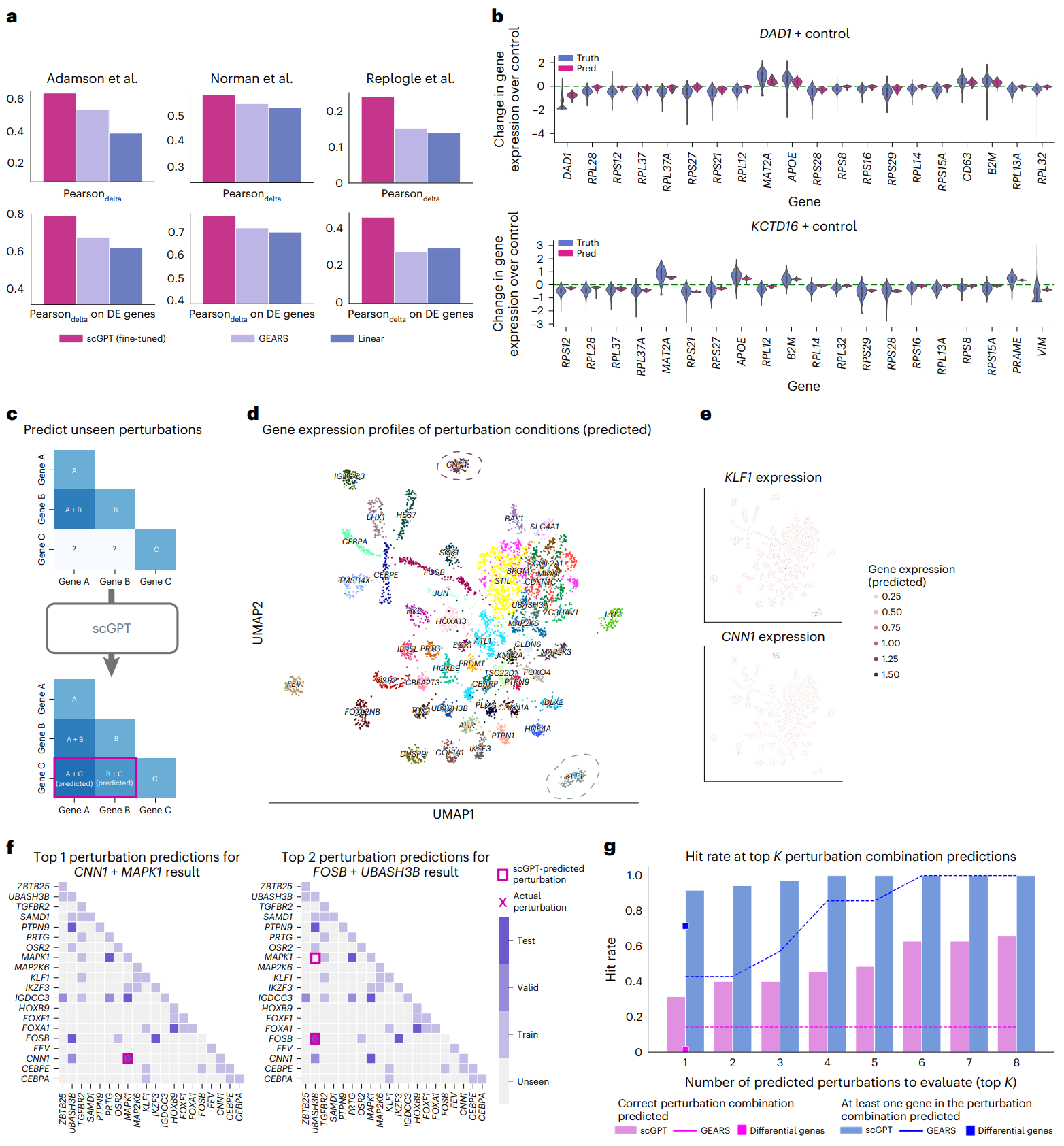
2.基因扰动响应和逆扰动的预测
3.多批次多模态数据整合

4.基因token嵌入分析
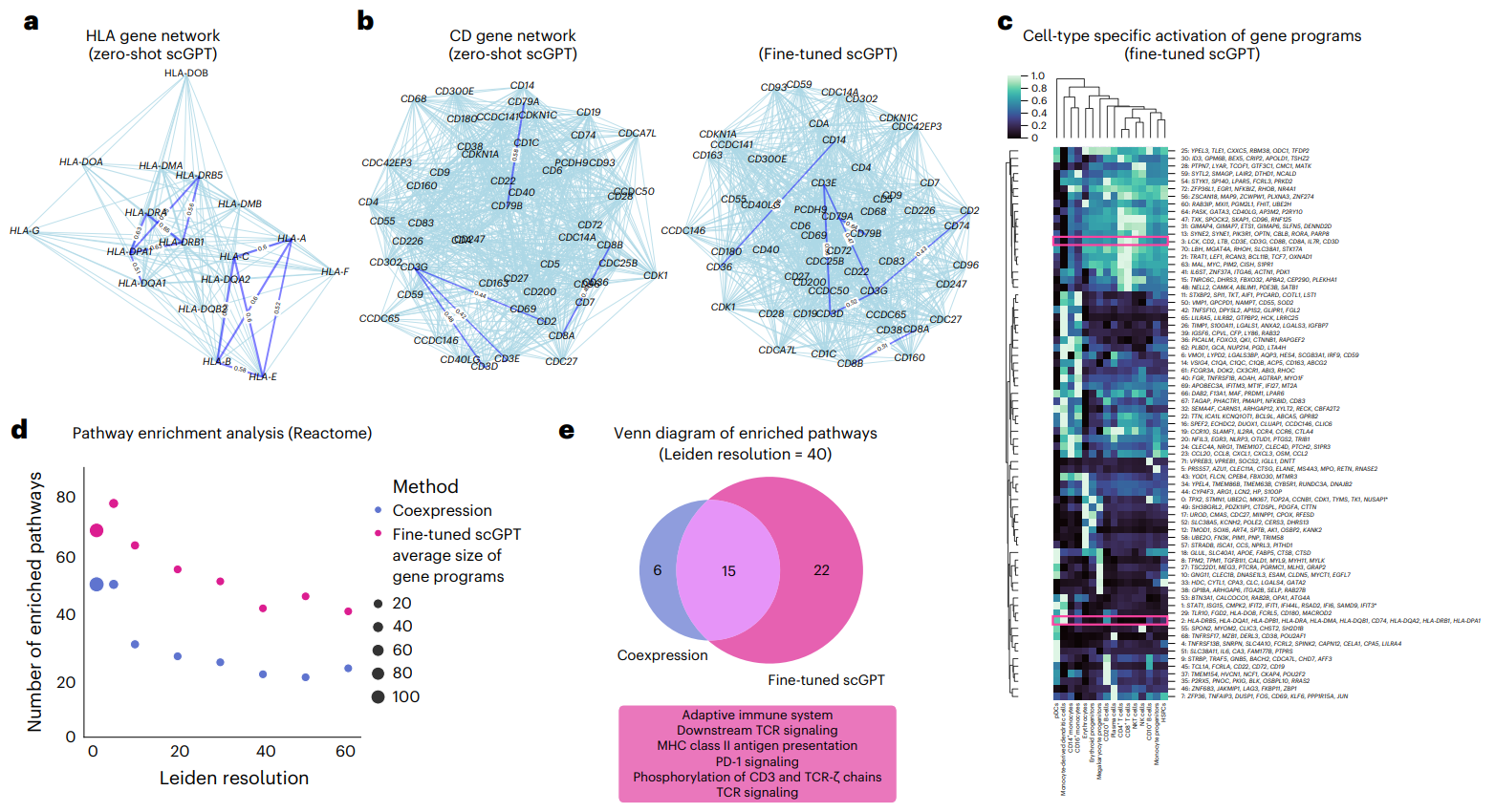
5.基于注意力的基因互作分析