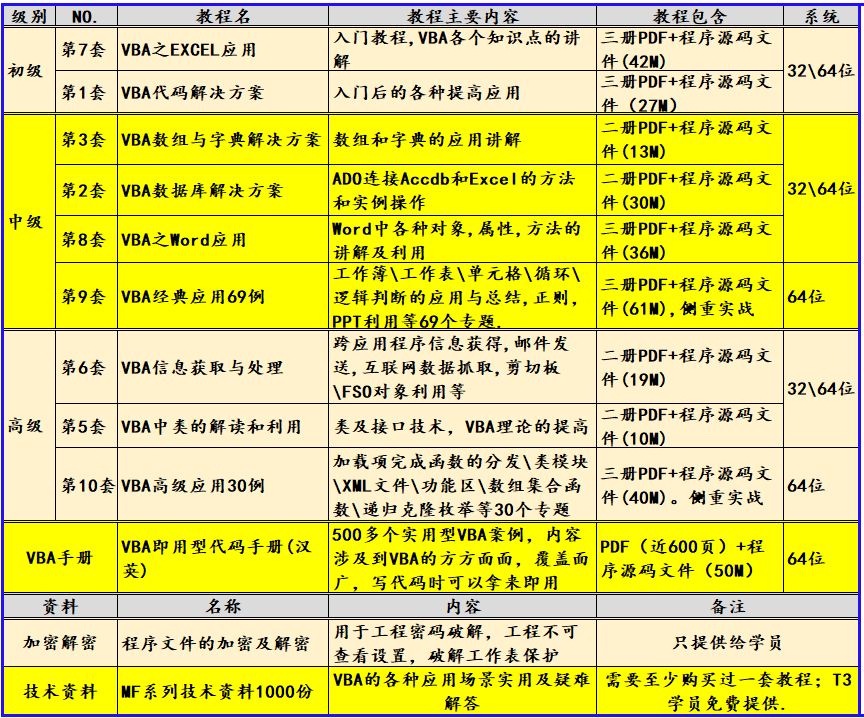
起源是由于bam文件没有过滤完全,导致calling出来的vcf文件还有线粒体中的染色体存在,因为在金标准文件中只有1-22号和X染色体,不包含线粒体和Y染色体,因为我使用的金标准文件是来自NA12878,是一位白种人女性。因此,vcf文件需要进一步过滤。
1、使用vcftools软件提取特定染色体
要使用vcftools提取VCF文件中chr1染色体上的所有变异,需要遵循vcftools的命令行选项来指定输入文件、染色体编号以及输出文件的名称
如果你的VCF文件是压缩的(通常以.vcf.gz结尾),你可以使用--gzvcf选项;如果文件未压缩,则使用--vcf选项。
vcftools --vcf bcftools.vcf --chr chr1 --recode --recode-INFO-all --out chr1_output--recode告诉vcftools将筛选后的变异重新编码为VCF格式
--recode-INFO-all确保在输出文件中包含所有INFO字段的信息
--out chr1_output指定了输出文件的前缀(vcftools会自动添加.recode.vcf作为后缀来创建最终的VCF文件)。
![]()
输出文件有以上这些。
但是需要注意的是,--chr 后面是什么取决于你的vcf文件里CHROM这一列是什么命名的。
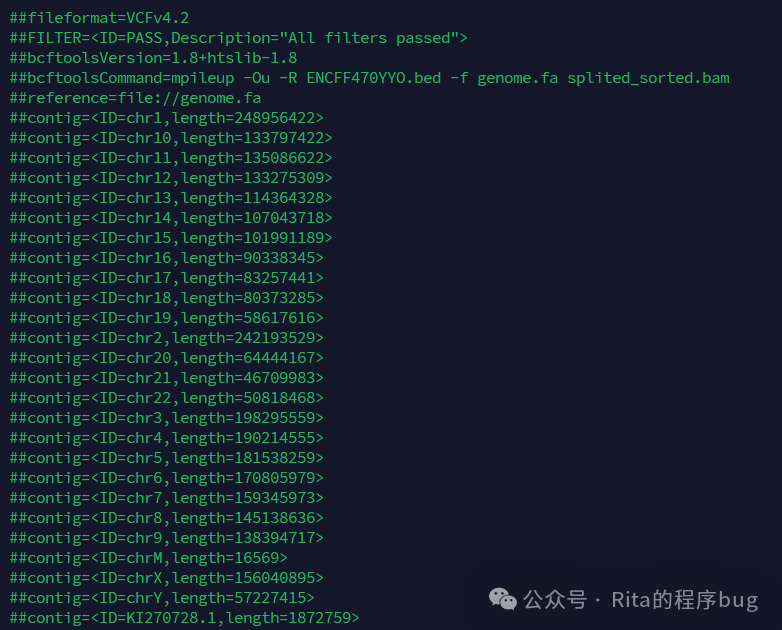
我的bcftools.vcf头文件长这样![]()
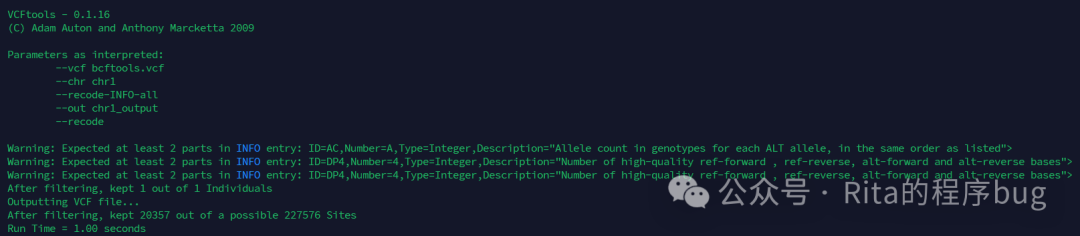
还是挺快的

最后我将chr1-22,和chrX染色体提取了出来
以.log结尾的都是日志文件
2、使用bcftools软件对多个vcf文件进行合并
一个常用的工具是bcftools concat(来自bcftools包,它是samtools的配套工具集),它专门用于合并多个VCF文件。
但是,请注意,在合并VCF文件之前,你需要确保这些文件在基因组位置上是兼容的(即它们应该覆盖相同的基因组区域或至少是互不重叠的),并且它们的INFO和FORMAT字段应该是一致的(或者至少是你想要合并的那些字段)。
bcftools concat -O v -o merged.vcf chr1_output.recode.vcf chr2.recode.vcf chr3.recode.vcf chr4.recode.vcf chr5.recode.vcf chr6.recode.vcf chr7.recode.vcf chr8.recode.vcf chr9.recode.vcf chr10.recode.vcf chr11.recode.vcf chr12.recode.vcf chr13.recode.vcf chr14.recode.vcf chr15.recode.vcf chr16.recode.vcf chr17.recode.vcf chr18.recode.vcf chr19.recode.vcf chr20.recode.vcf chr21.recode.vcf chr22.recode.vcf chrX.recode.vcf输出文件为
![]()
![]()
-
bcftools concat是用于合并VCF文件的命令。 -
-O v来明确指定输出格式为VCF,这是默认设置 -
-Oz选项指定输出文件应该被压缩(使用gzip),并且输出格式为VCF(默认是BCF,但VCF也受支持,尽管没有特定的选项来明确指定)。 -
-o merged.vcf.gz指定输出文件的名称。 -
后面跟着的是要合并的VCF文件列表