WGCNA(Weighted Gene Co-expression Network Analysis,加权重基因共表达网络分析)
WGCNA是一种用于分析基因表达数据的系统生物学方法。主要用于识别在基因表达数据中呈现共表达模式的基因模块,并将这些模块与样本特征(如临床特征、表型数据)相关联,进而识别关键驱动基因或生物标志物。

这里需要置入几个推文链接,因为笔者认为这几位老师写的内容已经把WGCNA的基础知识详细地展示了出来。
①生信技能树:https://mp.weixin.qq.com/s/3w2e6LwRowMsm2ZzyMAvdA
②生信菜鸟团:https://mp.weixin.qq.com/s/pLIGZhopfi-NCNtSuvwOBg
③生信星球:https://mp.weixin.qq.com/s/VlACgl6xlQ9YON4153Rp9g
④生信小白要知道:https://mp.weixin.qq.com/s/6atAeTKzPtIvCdMmWPIJEw
笔者也没有信心能够超越老师们的内容,详细解释可直接点击上述链接进行阅读。
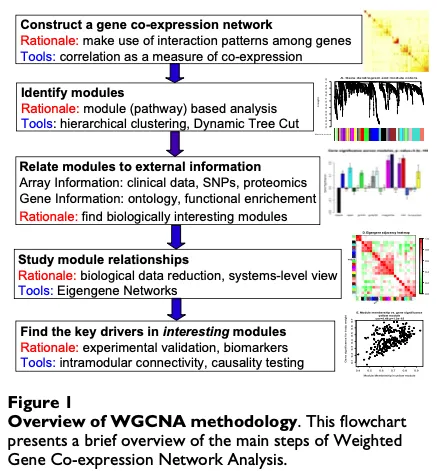
这是WGCNA分析的流程:基因共表达网络 — 识别基因模块 — 关联基因模块与表型 — 研究基因模块间关系 — 从感兴趣的基因模块中寻找关键驱动基因
我们要注意并理解WGCNA分析的关键点,个人认为主要有以下几点:
1、构建无标度网络,识别关键模块基因。这里的无标度网络就像是社会中的人与人之间网络关系,大多数人之间的关系是普通的,但有少部分的人具有很强的"引力",与他们链接在一起的有一大帮子人。这里的少部分人在WGCNA中就相当于是关键模块基因,是非常重要的节点,如果没有了这个节点这个网络就可能会“瘫痪”。
2、在构建这个无标度网络的时候采用了加权共表达的方式,并且由定义的软阈值去计算加权网络。加权共表达的方式非常好理解,比如人与人之间的交流肯定存在一定的强弱关系,不会只是好与不好的正反两种情况。
3、软阈值的作用是为了更好的放大或者缩小不同节点之间的相关性情况,从而减少在未使用软阈值情况下节点因为稍未达到阈值而被认为“不重要”的情况发生。
总之WGCNA可以采用更加优化的加权方式识别重要的模块及其基因。
分析步骤
1、导入数据
rm(list=ls())
library(WGCNA)
library(ggplot2)
load(file = "./data.Rdata")
dir.create("practice")
setwd("practice")
2、数据预处理
如果有批次效应,需要先进行去除;
如果数据存在系统偏移,需要进行quantile normalization;
标准化推荐使用DESeq2中的varianceStabilizingTransformation方法,
或将基因标准化后的数据(如FPKM、CPM等)进行log2(x+1)转化或者归一化后的芯片数据。
identical(colnames(exp),rownames(pd))
boxplot(exp)

3、提取表达矩阵并进行质控
datExpr0 = t(exp[order(apply(exp, 1, mad), decreasing = T)[1:5000],])
#datExpr0 = t(exp[order(apply(exp, 1, var), decreasing = T)[1:round(0.25*nrow(exp))],])
#datExpr0 = as.data.frame(t(exp))
datExpr0[1:4,1:4]
#rownames(datExpr0) = names(exp)[-c(1:8)]
# 基因和样本质控----
gsg = goodSamplesGenes(datExpr0, verbose = 3)
# `verbose` 参数被设置为 `3`,用于控制函数 `goodSamplesGenes` 的详细输出程度。
# `verbose = 0`:不产生任何输出,只返回结果,通常用于静默模式。
# `verbose = 1`:产生基本的信息输出,以提供一些关于函数执行进度的信息。
# `verbose = 2`:产生更详细的输出,可能包括一些中间步骤的信息。
# `verbose = 3`:产生最详细的输出,通常包括每个步骤的详细信息,用于调试和详细分析。
gsg$allOK # 返回TRUE则继续
if (!gsg$allOK){
# 把含有缺失值的基因或样本打印出来
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# 去掉那些缺失值
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
# 样本质控
sampleTree = hclust(dist(datExpr0), method = "average")
# 使用欧氏距离 (dist(dat)) 计算样本间的距离矩阵。
# 使用层次聚类方法(平均连接法)构建聚类树。
png(file = "2.sampleClustering.png", width = 10000, height = 2000,res = 300)
par(cex = 0.6)
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers",
sub="", xlab="", cex.lab = 1.5,cex.axis = 1.5, cex.main = 2)
#abline(h = 140, col = "red") #根据实际情况而定是否要划线去除离群点
dev.off()
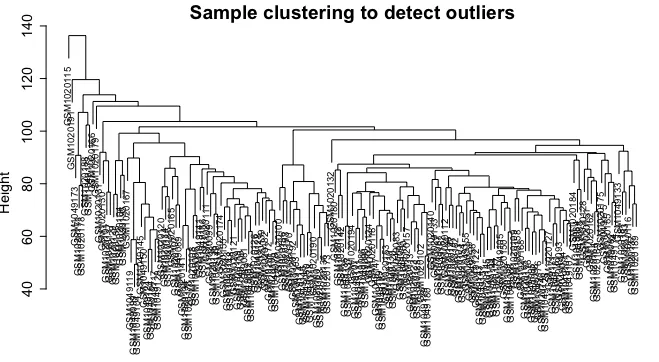
4.异常值筛选及质控
# 如果有异常值就需要去掉
if(F){
clust = cutreeStatic(sampleTree, cutHeight = 140, minSize = 10)
table(clust) # 0代表切除的,1代表保留的
keepSamples = (clust!=0)
datExpr = datExpr0[keepSamples, ]
}
# cutHeight = 200:用于指定在层次聚类树中切割的高度。在树状结构中,高度表示样本之间的相似性或距离。
# 通过指定 cutHeight,你可以控制在哪个高度水平切割树,从而确定最终的簇数。
# minSize = 10:用于指定最小簇的大小。在进行切割时,如果某个簇的大小小于 minSize,
# 则可能会合并到其他簇中,以确保生成的簇都具有足够的样本数。
# 切除完了之后需要再回到上面的代码进行做图!
#去除了异常样本之后需要跟临床信息做一个对应哦
#s <- intersect(rownames(datExpr),rownames(clinical))
#clinical <- clinical[s,]
#datExpr <- datExpr[s,]
#a <- identical(rownames(datExpr),rownames(clinical));a
#没有异常样本就不需要去除
datExpr = datExpr0
5、添加表型矩阵联合
# 这里要注意的是,用于关联分析的性状必须是数值型特征(比如年龄、体重、基因表达水平等)。
# 如果是分类变量(比如性别、地区等),则需要转换为0-1矩阵的形式
# (1表示属于此组或有此属性,0表示不属于此组或无此属性)。
library(stringr)
#搜索列名
tmp = data.frame(colnames(pd))
traitData = data.frame(
Female = as.numeric(grepl("F", pd$gender)),
Male = as.numeric(grepl("M", pd$gender))
)
str(traitData)
table(traitData[,1])
dim(traitData)
names(traitData)
datTraits = traitData
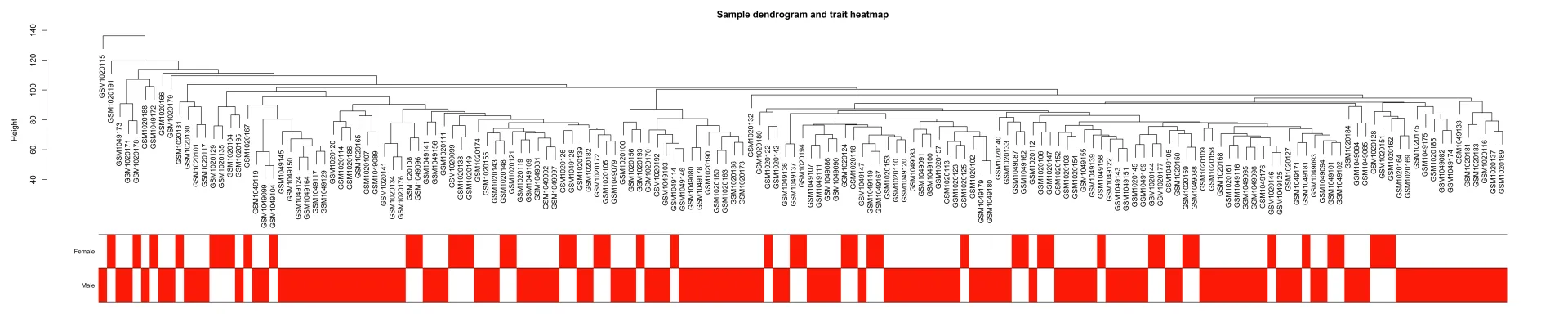
#表型和样本的相关性----
sampleTree2 = hclust(dist(datExpr), method = "average")
# 用颜色表示表型在各个样本的表现: 白色表示低,红色为高,灰色为缺失
traitColors = numbers2colors(datTraits, signed = FALSE)
# 把样本聚类和表型热图绘制在一起
png(file = "3.Sample dendrogram and trait heatmap.png", width = 10000, height = 2000,res = 300)
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
dev.off()
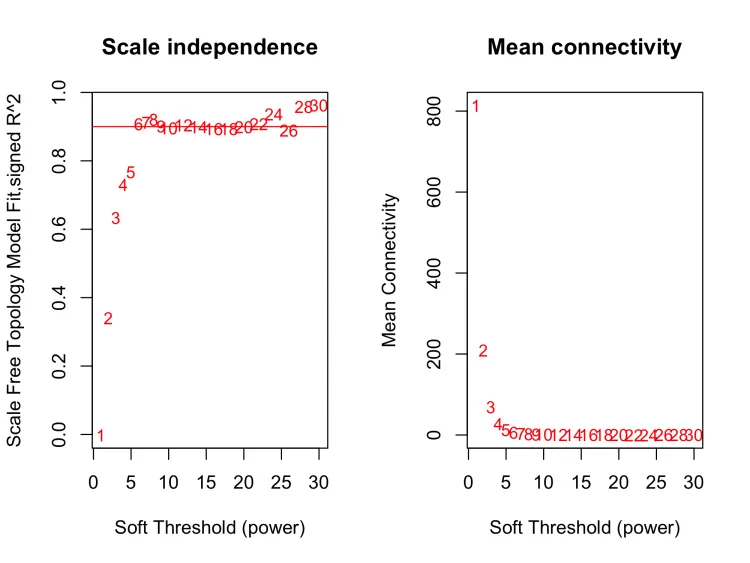
6、软阈值筛选
#设置一系列软阈值(默认1到30)
powers = c(1:10, seq(from = 12, to=30, by=2))
#帮助用户选择合适的软阈值,进行拓扑网络分析
#需要输入表达矩阵、设置的阈值范围、运行显示的信息程度(verbose=0不显示任何信息)
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
sft$powerEstimate
# pickSoftThreshold 函数的输出,用于选择最佳的软阈值以构建基因共表达网络或网络模型。
# powerEstimate:这是估计的最佳软阈值的值,它是一个整数。在这个结果中,估计的最佳软阈值是6。
# fitIndices:这是一个数据框,包含了不同软阈值下的拟合指标。每一行代表一个不同的软阈值(在 powers 中定义),列包括以下信息:
# Power:软阈值的幂次。
# SFT.R.sq:Scale Free Topology Model Fit,表示拟合模型的拟合度,通常用 R^2 衡量。较高的值表示模型更好地拟合数据。
# slope:表示软阈值幂次与拟合模型的斜率,通常用于衡量网络的无标度拓扑结构。一般来说,斜率绝对值越小,模型越能保持无标度拓扑结构。
# truncated.R.sq:表示截断模型的拟合度,也是一个衡量模型拟合程度的指标。
# mean.k.:平均连接度(Mean Connectivity),用于描述网络中节点的连接性。
# median.k.:中位数连接度,是平均连接度的中位数。
# max.k.:最大连接度,表示网络中具有最多连接的节点的连接数。
png(file = "4.Soft threshold.png", width = 2000, height = 1500,res = 300)
par(mfrow = c(1,2))
cex1 = 0.9
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",
ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"))
#注意这里的-sign(sft$fitIndices[,3])中sign函数,它把正数、负数分别转为1、-1
text(sft$fitIndices[,1],
-sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red")
# 设置筛选标准h=r^2^=0.9。这里的0.9是个大概的数,就是看左图软阈值6大概对应的位置
abline(h=cex1,col="red")
#看一下Soft threshold与平均连通性
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",
ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
dev.off()
6.1、经验power (无满足条件的power时选用)
# 无向网络在power小于15或有向网络power小于30内,没有一个power值可以使
# 无标度网络图谱结构R^2达到0.8,平均连接度较高如在100以上,可能是由于
# 部分样品与其他样品差别太大。这可能由批次效应、样品异质性或实验条件对表达影响太大等造成。可以通过绘制样品聚类查看分组信息和有无异常样品。
# 如果这确实是由有意义的生物变化引起的,也可以使用下面的经验power值。
if (is.na(power)){
power = ifelse(nSamples<20, ifelse(type == "unsigned", 9, 18),
ifelse(nSamples<30,
ifelse(type == "unsigned", 8, 16),
ifelse(nSamples<40, ifelse(type == "unsigned", 7, 14),
ifelse(type == "unsigned", 6, 12))
)
) }
7、一步法构建网络
sft$powerEstimate ##记得修改power
net = blockwiseModules(datExpr, power = 6,
TOMType = "unsigned",
minModuleSize = 30, #原来是30
reassignThreshold = 0,
mergeCutHeight = 0.25,#原来是0.25
deepSplit = 2 ,#原来是2
numericLabels = TRUE,
pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "testTOM",
verbose = 3)
# deepSplit 参数调整划分模块的敏感度,值越大,越敏感,得到的模块就越多;
# minModuleSize 参数设置最小模块的基因数,值越小,小的模块就会被保留下来;
# mergeCutHeight 设置合并相似性模块的距离,值越小,就越不容易被合并,保留下来的模块就越多。
# power = sft$powerEstimate:这里指定了软阈值的取值。
# sft$powerEstimate 就是我们之前计算得到的软阈值,它用来控制共表达网络的连接强度。
# maxBlockSize = nGenes:指定了最大的模块大小。在构建基因模块时,会将基因分成多个子模块,这个参数用来限制子模块的最大大小。
# nGenes 是基因的总数,通常用来作为最大模块大小的上限。(4G内存电脑可处理8000-10000个,16G内存电脑可以处理2万个,32G内存电脑可以处理3万个,计算资源允许的情况下最好放在一个block里面。)
# TOMType = "unsigned":指定了共表达网络的类型,这里设置为 "unsigned",表示构建无符号的网络。计算TOM矩阵时,是否考虑正负相关性,默认为"signed",但是根据幂律转换的邻接矩阵(权重)的非负性,所以我们认为这里选择"signed"也没有太多的意义。
# minModuleSize = 30:指定了每个模块的最小大小,也就是每个基因模块中至少包含多少个基因。
# reassignThreshold = 0:控制模块的重新分配。如果模块中的基因与其他模块的相关性低于这个阈值,它们可能会被重新分配到其他模块。
# mergeCutHeight = 0.25:用来合并基因模块。当两个模块的相关性超过这个阈值时,它们可能会被合并成一个更大的模块。越大得到的模块就越少。
# numericLabels = TRUE:表示模块将使用数值标签,返回数字而不是颜色作为模块的名字,后面可以再转换为颜色。
# pamRespectsDendro = FALSE:控制 PAM(Partitioning Around Medoids)聚类算法是否遵循树状结构。这里设置为 FALSE,表示不遵循树状结构。
# saveTOMs = F:用来控制是否保存共表达网络的拓扑重叠矩阵(TOM)。这里设置为 FALSE,表示不保存。最耗费时间的计算,有需要的话,大家存储起来,供后续使用。
# verbose = 3:用来控制输出的详细程度。设置为3可以输出较为详细的信息,用于跟踪函数的执行进程。
# corType:用来选择计算相关性的方法。默认“pearson”,还可以是“bicor”,“bicor”更能考虑离群点的影响bicor。
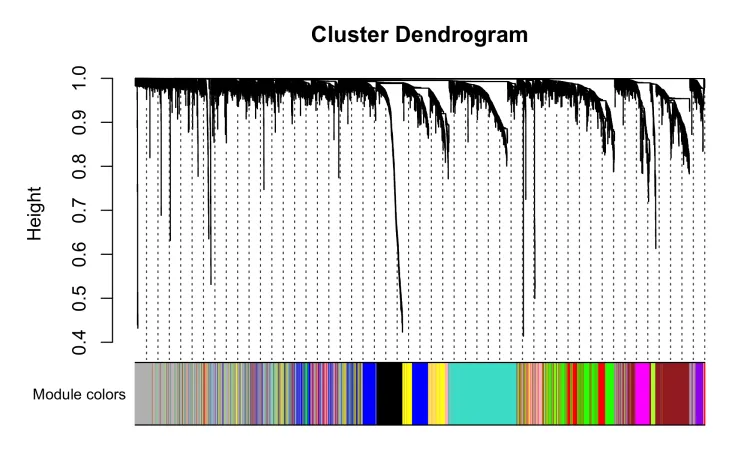
table(net$colors)
#模块可视化---
mergedColors = labels2colors(net$colors)
png(file = "5.DendroAndColors.png", width = 2000, height = 1200,res = 300)
plotDendroAndColors(net$dendrograms[[1]],
mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
dev.off()
8、获取模块基因
moduleLabels = net$colors # 对应多步法里面的mergedColors = merge$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs # 对应多步法里的mergedMEs = merge$newMEs
geneTree = net$dendrograms[[1]] #适用于一步法
g = merge(as.data.frame(table(net$colors)),
as.data.frame(table(mergedColors)))
g
genes = list()
for(i in 1:nrow(g)){
genes[[i]] = names(net$colors)[net$colors==g$Var1[[i]]]
}
names(genes) = g$mergedColors
save(genes,file = "genes.Rdata")
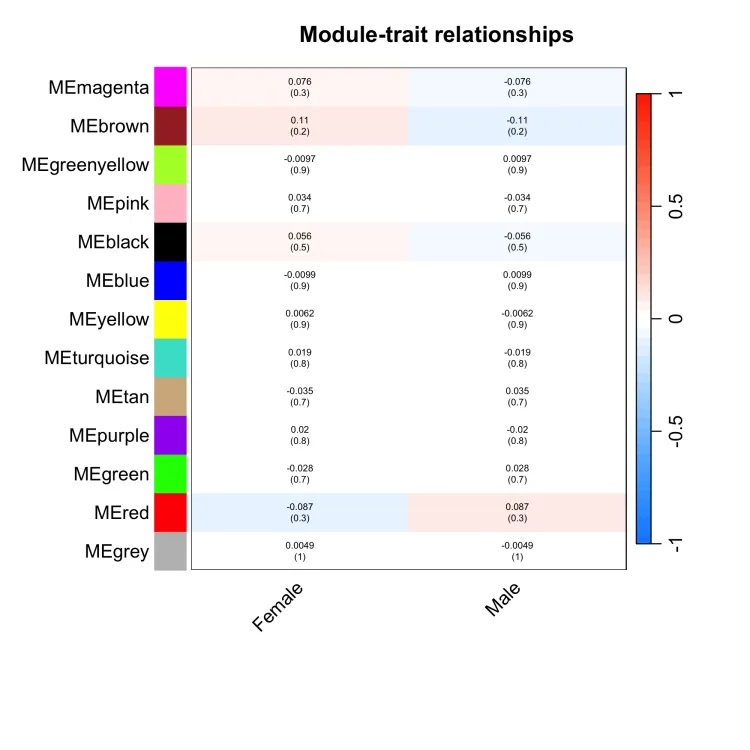
9、表型热图展示
# 得到基因、样本数量
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
# 用color labels重新计算MEs(Module Eigengenes:模块的第一主成分)
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
moduleTraitCor = cor(MEs, datTraits, use = "p") #(这是重点)计算ME和表型相关性
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
#热图
png(file = "6.labeledHeatmap.png", width = 2000, height = 2000,res = 300)
# 设置热图上的文字(两行数字:第一行是模块与各种表型的相关系数;
# 第二行是p值)
# signif 取有效数字
textMatrix = paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "")
dim(textMatrix) = dim(moduleTraitCor)
par(mar = c(8, 8.5, 3, 3))
# 然后对moduleTraitCor画热图
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = blueWhiteRed (50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
dev.off()
10、提取module信息
# 把各个module的名字提取出来(从第三个字符开始),用于一会重命名
modNames = substring(names(MEs), 3)
# 得到矩阵
geneModuleMembership = as.data.frame(cor(datExpr, MEs, use = "p"))
# 矩阵t检验
MMPvalue = as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples))
# 修改列名
names(geneModuleMembership) = paste("MM", modNames, sep="")
names(MMPvalue) = paste("p.MM", modNames, sep="")
11、提取表型信息
# 先将感兴趣表型instrait提取出来,用于计算矩阵
i = 2
assign(colnames(traitData)[i],traitData[i])
# 得到矩阵
instrait = eval(parse(text = colnames(traitData)[i]))
geneTraitSignificance = as.data.frame(cor(datExpr, instrait, use = "p"))
# 矩阵t检验
GSPvalue = as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples))
# 修改列名
names(geneTraitSignificance) = paste("GS.", names(instrait), sep="")
names(GSPvalue) = paste("p.GS.", names(instrait), sep="")
12、合并两个相关性矩阵,找到和模块、表型都高度相关的基因
module = "red"
#module = "turquoise"
png(file = paste0("7.MM-GS_red.png"), width = 2000, height = 2000,res = 300)
column = match(module, modNames) #找到目标模块所在列
moduleGenes = moduleColors==module #找到模块基因所在行,因为这里的颜色和基因的位置是对应的
par(mfrow = c(1,1))
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = paste("Gene significance for",names(datTraits)[i]),
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
dev.off()
13、导出相关信息
write.csv(geneModuleMembership,"geneModuleMembership.csv")
write.csv(geneTraitSignificance,"geneTraitSignificance.csv")
genes1 = as.data.frame(genes$red)
write.csv(genes1,file = "genes_red.csv")
参考资料:
1、WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008 Dec 29:9:559.
2、WGCNA: https://github.com/jmzeng1314/my_WGCNA
3、小杜的生信笔记: https://mp.weixin.qq.com/s/m441hz17sUFvT5g3nBoC_Q
4、生信小白要知道: https://mp.weixin.qq.com/s/fCvLizKQNWDQKeWBuSG3UQ
5、Biomamba 生信基地: https://mp.weixin.qq.com/s/5OUY5KDwgi05MlFrV_7Qjw
6、生信技能树:
https://mp.weixin.qq.com/s/HDCbJR4nfLlzeaDJmYO_gA https://mp.weixin.qq.com/s/3w2e6LwRowMsm2ZzyMAvdA https://mp.weixin.qq.com/s/9vEkaXZvoaoCdf_t8S1aBA
7、生信星球: https://mp.weixin.qq.com/s/T14K1FWkpTtIhWlcUSrK-A
致谢:感谢曾老师、小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -