探索疾病的免疫微环境是当下研究的热点,因此这也要求我们对免疫浸润分析的各种工具要有所了解,这些工具包括: CIBERSORT,quanTIseq,EPIC (这三种方法可得到不同细胞亚群的百分比结果/绝对丰度), xCell,TIMER,MCPcounter,ImmuCellAI (这四种方法可得到相对丰度),ESTIMATE (用于进行基质、免疫、Estimate和肿瘤纯度评分)等。
1. CIBERSORT
原理: 基于支持向量回归(SVR)的一种去卷积算法,用于通过转录组数据估算复杂组织样本中细胞类型的组成。使用预定义的参考基因表达特征矩阵来推断各个细胞类型的比例。Cibersort最终得到的值是百分比,可以比较同一样本中不同细胞群体的比例,也可以比较不同样本中不同细胞群体的比例。
适用场景: 适用于从基因表达数据中推断不同细胞类型的相对比例,尤其适合肿瘤样本。
2. quanTIseq
原理: 基于混合线性模型,通过基因表达数据去卷积来估算免疫细胞类型的组成。quanTIseq提供了针对多种细胞类型的绝对量估算。quantTIseq最终得到的值是百分比,可以比较同一样本中不同细胞群体的比例,也可以比较不同样本中不同细胞群体的比例。
适用场景: 适合需要定量估算多个免疫细胞类型绝对丰度的应用场景,特别是在肿瘤免疫研究中。
3. EPIC
原理: 通过参考基因表达矩阵去卷积来估算细胞类型的组成,特别关注肿瘤和免疫细胞在组织样本中的比例。EPIC还能估计肿瘤细胞的比例。EPIC最终得到的值是百分比,可以比较同一样本中不同细胞群体的比例,也可以比较不同样本中不同细胞群体的比例。
适用场景: 适用于分析肿瘤样本中免疫细胞和肿瘤细胞的比例,尤其在涉及肿瘤异质性研究时。
4. xCell
原理: 通过扩展的ssGSEA方法,整合了细胞类型特异性基因集来推断免疫细胞的浸润水平。xCell基于大量参考基因表达数据,并考虑了样本内的基因表达变异。xCell最终得到的值是相对丰度估计,该方法可比较同一细胞类型在不同样本中的丰度,但不能用于同一样本里不同细胞类型之间的丰度比较。
适用场景: 对需要从转录组数据中进行广泛免疫细胞类型注释的研究非常有用。
5. MCPcounter
原理: 基于特异性标记基因集合,定量评估复杂样本中多个免疫细胞和非免疫细胞的相对丰度。MCPcounter通过对这些标记基因的表达量进行加权和求和来估算细胞丰度。MCPcounter最终得到的值是相对丰度估计,该方法可比较同一细胞类型在不同样本中的丰度,但不能用于同一样本里不同细胞类型之间的丰度比较。
适用场景: 适用于希望得到肿瘤微环境中多个细胞类型的绝对定量评估的研究。
6. ImmuCellAI
原理: 主要基于基因表达数据,用于评估24种免疫细胞类型的浸润水平。ImmuCellAI特别关注T细胞亚群的推断。ImmuCellAI最终得到的值是相对丰度,该方法可比较同一细胞类型在不同样本中的丰度,但不能用于同一样本里不同细胞类型之间的丰度比较。
适用场景: 在T细胞相关的免疫研究中表现突出,特别适用于需要详细T细胞亚群分析的应用场景。
7. TIMER
原理: 使用统计模型来推断肿瘤样本中的6种主要免疫细胞类型的浸润水平。TIMER结合了肿瘤纯度的估计,并考虑了肿瘤细胞与免疫细胞之间的相互作用。TIMER最终得到的值是相对丰度,该方法可比较同一细胞类型在不同样本中的丰度,但不能用于同一样本里不同细胞类型之间的丰度比较。
适用场景: 专门针对肿瘤免疫微环境的研究,特别是当肿瘤样本中的肿瘤纯度可能影响分析结果时。
8.ESTIMATE
原理: 通过基因表达特征推断肿瘤样本中的肿瘤纯度以及免疫细胞和基质细胞的比例。ESTIMATE主要使用特定的基因表达特征(本质上是ssGSEA)来评估样本的免疫评分和基质评分,基于此进一步推断肿瘤纯度。ESTIMATE最终得到的值是相对丰度,该方法可比较同一细胞类型在不同样本中的丰度,但不能用于同一样本里不同细胞类型之间的丰度比较。
适用场景: 特别适合用于评估肿瘤样本中的免疫和基质成分,并推断肿瘤纯度。
在19年的发表在Bioinformatics期刊上的一篇文献中,作者对比了其中几种工具。
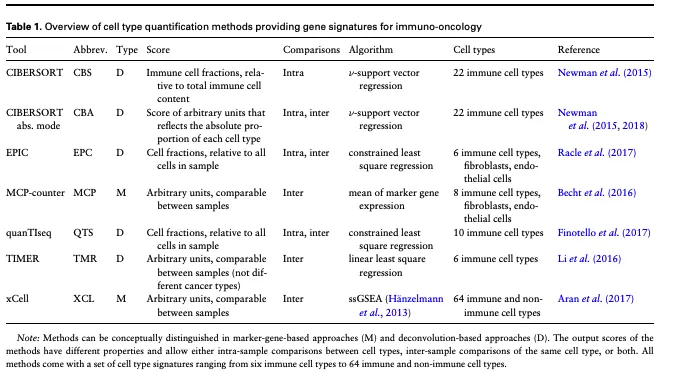
这些方法通常可以分为两类:基于标记基因的方法和基于去卷积的方法。
● 基于标记基因的方法:这种方法使用一组特定的基因,这些基因是某种细胞类型的特征。这些基因集合通常来源于对每种免疫细胞类型的靶向转录组学研究,或者是通过全面的文献查找和实验验证得出的。通过使用异质样本中这些标记基因的表达值,这些模型可以独立地量化每种细胞类型,有的会将这些量化结果聚合成一个丰度评分(如MCP-counter,Becht等,2016),或者通过对标记基因的富集进行统计检验(如xCell,Aran等,2017)。
● 基于去卷积的方法:这种方法是使用算法公式,描述样本的基因表达是混合细胞类型表达谱的加权和。通过求解这个逆问题(inverse problem),可以在给定特征矩阵和大量基因表达的情况下推断出细胞类型比例。在实际操作中,这个问题可以通过线性最小二乘回归(如TIMER,Li等,2016)、约束最小二乘回归(如quanTIseq和EPIC,Finotello等,2017;Racle等,2017),或者ν-支持向量回归(如CIBERSORT,Newman等,2015)来解决。

研究者根据三个标准去评估了不同细胞类型适用的方式: 整体表现: 表示该方法预测的细胞比例与基准中的已知比例的吻合程度。绝对评分: 该方法提供的评分可以解释为细胞的实际比例。如果某方法不提供绝对评分,则它只能支持在同一实验数据集中进行样本间的比较,即这个评分仅在与其他样本进行比较时有意义。背景预测: 表示该方法是否倾向于预测出一种实际不存在的细胞类型。
研究者认为EPIC和quanTIseq是唯一提供“绝对评分”的方法,这种评分代表了细胞的"实际"(我认为是相对的实际)比例。其他方法则提供的是相对评分,且仅在与同一数据集中的其他样本进行比较时才有意义。基于这一点以及它们在整体表现上的稳健性,研究者们推荐EPIC和quanTIseq作为通用去卷积的首选方法。
在实际应用中,绝对评分并不总是必要的。例如,在临床试验中,可以使用相对评分方法来推断治疗组与对照组之间的倍数变化,或者监测纵向样本中免疫成分的变化。在这种情况下,由于MCPcounter具有高度特异性的标记基因,并在交叉污染测试中表现优异,它是一个很好的选择。
去卷积方法的一个局限性是它们容易受到背景预测的影响,即会预测出实际不存在的细胞类型的比例(比例很小,但是存在) 。因此,如果关注某种细胞类型的存在或不存在,建议使用xCell。该文章最后没有在推荐里提到cibersort 。
小结:
1. 算法的使用:
● CIBERSORT、quanTIseq、EPIC 都使用了去卷积算法来估算样本中不同细胞类型的比例。
● MCP-counter 通过标记基因集合进行加权和求和,而不是传统的去卷积算法。
● TIMER 和 ESTIMATE 使用了基于统计模型和 ssGSEA 的方法来推断细胞类型的相对丰度。
2. 结果表示: ● CIBERSORT、quanTIseq、EPIC 产生的结果是百分比形式,可以直接用于比较不同细胞群体的比例。 ● xCell、MCPcounter、ImmuCellAI、TIMER、ESTIMATE 的结果是相对丰度估计,通常用于比较同一细胞类型在不同样本中的丰度。
3. 适用场景:
● CIBERSORT、EPIC 和 quanTIseq 适合复杂组织样本的分析,尤其肿瘤样本。
● xCell 和 ImmuCellAI 适合广泛免疫细胞类型注释和T细胞相关研究。
● TIMER 专注于肿瘤免疫微环境,特别考虑了肿瘤纯度对结果的影响。
● MCPcounter 提供了多个细胞类型的绝对定量估计,适用于多细胞类型分析。
● ESTIMATE 用于推断肿瘤纯度和分析肿瘤样本中的免疫和基质成分。
总而言之,个人认为无论是哪种方式都是运用数理方式进行模拟运算,得到最终的结果。对于医学/生命科学专业(非侧重生信)出身的科研者来说可以简单粗暴的全部整合分析一遍,综合最后的结果去判断。因为如果要细究算法原理,那首先需要判断数据的情况,这会给是医学/生命科学专业的伙伴们增加了更多的负担。并且模拟的数据和真实情况一定是百分百一致没有偏差吗,那必然不是这样的。所以每种方法大概的原理和知识确实是需要去了解,但不要刻意的去纠结,在满足适用性的前提下(大原则不能错),多种方法整合分析可能是一个较好的选择。
分析流程
分别展示Cibersot,quanTIseq,EPIC,xCell,MCP-counter,ESTIMATE。 而TIMER和ImmuCellAI更常用的是它们的网页工具。
1.导入数据并检查(整理好的TCGA肝癌数据)
rm(list = ls())
# 整理好的TCGA肝癌数据
load("TCGA-LIHC_sur_model.Rdata")
head(exprSet)[1:4,1:4]
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A
# WASH7P 1.913776 1.2986076 1.967382 1.586170
# AL627309.6 3.129116 0.5606928 3.831265 1.363539
# WASH9P 2.490476 2.8140204 2.960338 2.106464
# MTND1P23 2.773335 3.4114257 2.591028 3.353850
head(meta)
# ID OS OS.time race age gender stage T N M
# TCGA-FV-A495-01A TCGA-FV-A495-01A 0 0.03333333 WHITE 51 FEMALE II 2 NA 0
# TCGA-ED-A7PZ-01A TCGA-ED-A7PZ-01A 0 0.20000000 ASIAN 61 MALE II 2 NA 0
# TCGA-ED-A97K-01A TCGA-ED-A97K-01A 0 0.20000000 ASIAN 54 MALE III 3 0 0
# TCGA-ED-A7PX-01A TCGA-ED-A7PX-01A 0 0.20000000 ASIAN 48 FEMALE II 2 NA 0
# TCGA-BC-A3KF-01A TCGA-BC-A3KF-01A 0 0.26666667 WHITE 66 FEMALE I 1 NA 0
# TCGA-DD-A4NR-01A TCGA-DD-A4NR-01A 1 0.30000000 WHITE 85 FEMALE I 1 0 0
identical(rownames(meta),colnames(exprSet))
# [1] TRUE
group <- factor(meta$gender,levels = c("FEMALE","MALE"))
table(group)
# FEMALE MALE
# 119 246
# 用对性别进行了因子化处理,后面会用到
2、Cibersot
制作输入文件,表达矩阵文件输入要求:1.不可以有负值和缺失值; 2.不要取log; 3.如果是芯片数据,昂飞芯片使用RMA标准化,Illumina 的Beadchip 和Agilent的单色芯片,用limma处理。 4.如果是RNA-Seq表达量,使用FPKM和TPM都可以。需要把行名(基因名)变成其中一列
R包中内置了22种免疫细胞的基因表达特征数据(LM22.txt),包括了7种T细胞类型、naive和记忆B细胞、浆细胞、NK细胞和骨髓亚群
library(tidyverse)
exp = exprSet
exp2 = as.data.frame(exp)
exp2 = rownames_to_column(exp2)
write.table(exp2,file = "exp.txt",row.names = F,quote = F,sep = "\t")
f = "ciber_LIHC.Rdata"
if(!file.exists(f)){
#devtools:: install_github ("Moonerss/CIBERSORT")
library(CIBERSORT)
lm22f = system.file("extdata", "LM22.txt", package = "CIBERSORT")
ciber.results = cibersort(lm22f,
"exp.txt" ,
perm = 1000,
QN = T)
save(ciber.results,file = f)
}
load(f)
ciber.results[1:4,1:4]
ciber_re <- ciber.results[,-(23:25)]
row_sums <- rowSums(ciber_re)
head(row_sums)
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A TCGA-BC-A3KF-01A TCGA-DD-A4NR-01A
# 1 1 1 1 1 1

最终结果会有额外的三列,一般会去掉。
● P-value: 用来展示去卷积的结果在所有细胞类群中是否具有差异
● Correlation:参考矩阵与输入矩阵的特征基因相关性
● RMSE: Root mean squared error,参考矩阵与输入矩阵的特征基因标准差
并且可以看到row_sums行求和的内容,每一行代表一个样本,里边的22中免疫细胞的总和为1。
3、quanTIseq
quanTIseq开发的初衷是针对RNA-seq数据,并最终对10种免疫细胞进行绝对分数定量,最终得到每一个样本中不同免疫细胞之间的百分比。输入的数据为TPM格式。
library(quantiseqr)
quanTIseq_re <- run_quantiseq(expression_data = exprSet,
signature_matrix = "TIL10",
is_arraydata = FALSE,
is_tumordata = TRUE,
scale_mRNA = TRUE,
column = "gene_symbol")
head(quanTIseq_re)[1:4,1:4]
# Sample B.cells Macrophages.M1 Macrophages.M2
# TCGA-FV-A495-01A TCGA-FV-A495-01A 0.008236479 0.020688682 0.041672718
# TCGA-ED-A7PZ-01A TCGA-ED-A7PZ-01A 0.007310874 0.004809912 0.009260324
# TCGA-ED-A97K-01A TCGA-ED-A97K-01A 0.014541128 0.043945421 0.051301815
# TCGA-ED-A7PX-01A TCGA-ED-A7PX-01A 0.008072784 0.030297861 0.046511408
a <- quanTIseq_re[,-1]
row_sums <- rowSums(a)
head(row_sums)
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A TCGA-BC-A3KF-01A TCGA-DD-A4NR-01A
# 1 1 1 1 1 1
expression_data:tpm表达矩阵;signature_matrix:代表参考细胞类型的矩阵。包的内置数据,使用"TIL10"是对10种免疫细胞的计算。is_arraydata:是否为微阵列数据。is_tumordata:是否来自肿瘤样本。scale_mRNA:指示是否对表达数据进行标准化。
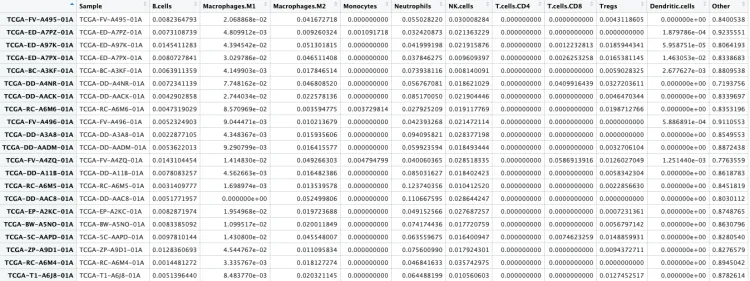
并且可以看到row_sums行求和的内容,每一行代表一个样本,里边的10种免疫细胞及other列的总和为1。
4、EPIC
EPIC 整合了多种主要免疫和其他非恶性细胞类型的参考基因表达谱,并将其作为参考模型,而bulk RNA-Seq数据则是在这种参考模型的基础上进行分析。开发者在github的vignettes中提到需要用TPM或者RPKM数据进行分析。
library(EPIC)
EPIC_raw <- EPIC(bulk = exprSet)
EPIC_re<-EPIC_raw$mRNAProportions
head(EPIC_re)[1:4,1:4]
# Bcells CAFs CD4_Tcells CD8_Tcells
# TCGA-FV-A495-01A 0.031375388 0.01912478 0.12230719 0.05138005
# TCGA-ED-A7PZ-01A 0.011843734 0.01169971 0.04835501 0.02911673
# TCGA-ED-A97K-01A 0.019552109 0.01682554 0.15680554 0.05896725
# TCGA-ED-A7PX-01A 0.009935306 0.01987824 0.09758243 0.04574178
row_sums <- rowSums(EPIC_re)
head(row_sums)
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A TCGA-BC-A3KF-01A TCGA-DD-A4NR-01A
# 1 1 1 1 1 1
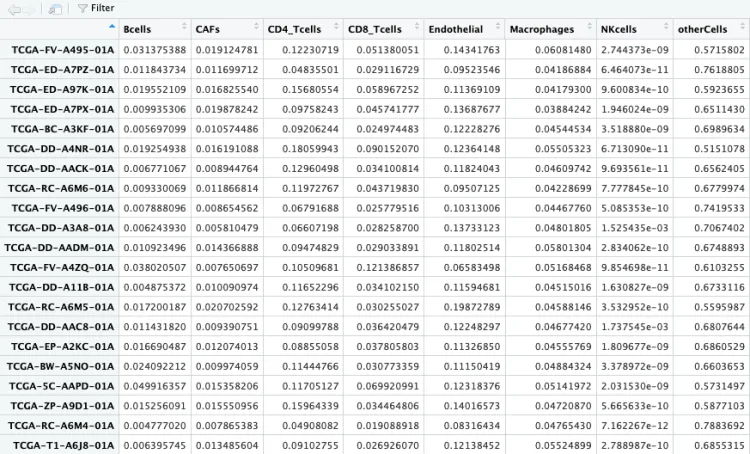
并且可以看到row_sums行求和的内容,每一行代表一个样本,里边的10种免疫细胞及other列的总和为1。
5、xCell
xCell使用的表达矩阵,行名基因、列名样本。其需要输入RPKM/FPKM/TPM/RSEM值,不可使用counts值。
xCell评分是基于signatures的富集分数,它与真正的细胞比例有线性相关性,但是不能把xCell评分作为细胞比例值。xCell可以在不同样本之间对比同一细胞类型的得分,但是不可在同一样本内比较不同细胞类型的得分。
library(xCell)
xCell_re <- xCellAnalysis(exprSet)
xCell_re[1:5,1:5]
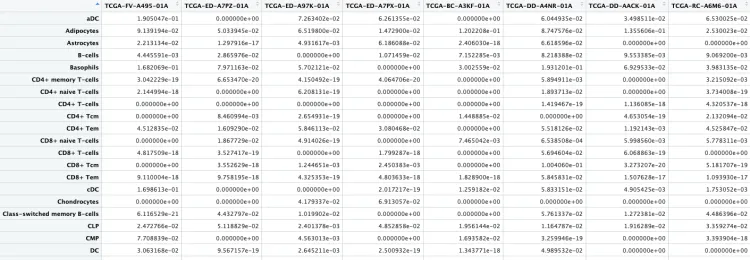
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A TCGA-BC-A3KF-01A
# aDC 0.190504736 0.000000e+00 0.072634025 0.06261355 0.000000e+00
# Adipocytes 0.091391941 5.033945e-02 0.065197997 0.01472900 1.202208e-01
# Astrocytes 0.022131342 1.297916e-17 0.004931617 0.06186088 2.406030e-18
# B-cells 0.004445591 2.865976e-02 0.000000000 0.01071459 7.152285e-03
# Basophils 0.168206883 7.971163e-02 0.057021215 0.00000000 3.002559e-02
6、MCPcounter
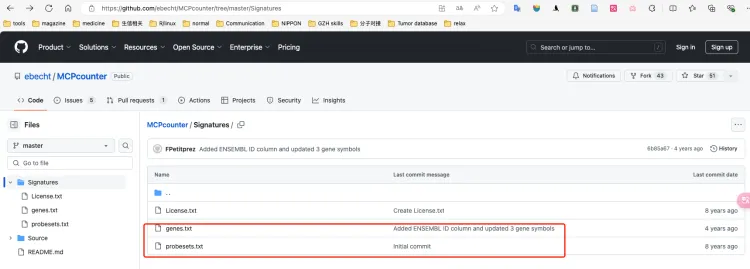
需要在该网站上https://github.com/ebecht/MCPcounter/tree/master/Signatures下载genes.txt和probesets. txt文件。现在运行的时候不下载也可以直接默认运行也行,但是需要把代码中的probesets和genes删去。
该方法是使用了芯片数据作为先验数据建立了模型,使用者输入数据最理想是使用芯片数据,网上很多教程里面有说使用TPM或者log2TPM均可。
MCPcounter使用线性模型来将基因表达量转化为特定类型免疫细胞的数量。这个模型的参数是在已知的免疫相关基因和对应免疫细胞数量的基础上进行训练得到的。经过模型转化后,得到了每种免疫细胞类型的数量估计值。这些值代表了该类型免疫细胞在样本中的丰度。
library(MCPcounter)
probesets <- data.table::fread("probesets.txt",data.table = F,header = F)
genes <- data.table::fread("genes.txt",data.table = F)
MCP_counter_re <- MCPcounter.estimate(exprSet,featuresType = "HUGO_symbols",
probesets=probesets,
genes=genes)
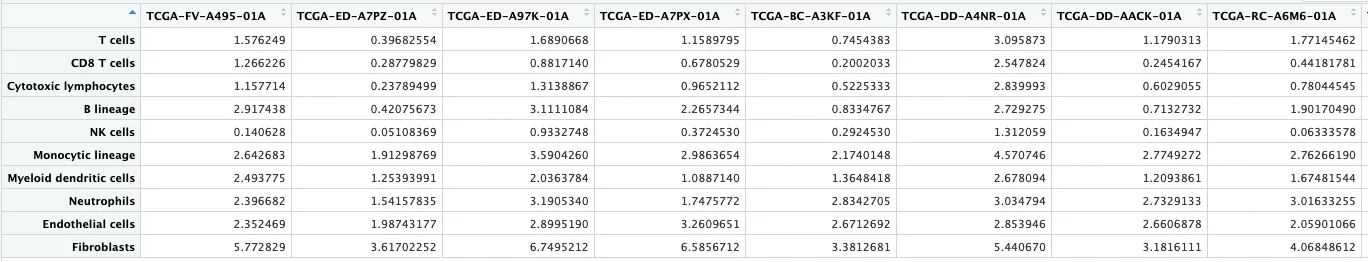
MCP_counter_re[1:5,1:5]
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A TCGA-BC-A3KF-01A
# T cells 1.576249 0.39682554 1.6890668 1.1589795 0.7454383
# CD8 T cells 1.266226 0.28779829 0.8817140 0.6780529 0.2002033
# Cytotoxic lymphocytes 1.157714 0.23789499 1.3138867 0.9652112 0.5225333
# B lineage 2.917438 0.42075673 3.1111084 2.2657344 0.8334767
# NK cells 0.140628 0.05108369 0.9332748 0.3724530 0.2924530
7、ESTIMATE
利用预筛选的基质/免疫相关基因集,使用基因表达数据预测肿瘤组织中基质/免疫细胞浸润水平,可以生成三或者四个分数: 基质评分(stromal score),评估肿瘤组织中的基质细胞浸润水平;免疫评分(immune score),评估肿瘤组织中的免疫细胞浸润水平;两个分数相加即得到 estimate score,可以用于推断肿瘤纯度(Tumor Purity)
ESTIMATE输入logCPM、logTPM与logFPKM格式的数据都可以,行为基因名,列为样本。另外,转录组测序数据与芯片数据计算过程的主要不同是platform选项修改为illumina。
library(estimate)
# 把表达矩阵转换成txt文件
write.table(exprSet, file = "exprSet.txt", sep = "\t", row.names = T, col.names = TRUE, quote = FALSE)
# 用内置函数把txt文件转换成gct文件
filterCommonGenes(input.f = "exprSet.txt",
output.f = "lihc_genes.gct",
id = "GeneSymbol") #或者使用EntrezID
# [1] "Merged dataset includes 8283 genes (2129 mismatched)."
# 进行estimateScore打分
estimateScore(input.ds = "lihc_genes.gct",
output.ds = "lihc_estimate_score.gct",
platform = "illumina") # 转录组需要用illumina平台,而芯片用affymetrix,除此之外还有agilent。
# [1] "1 gene set: StromalSignature overlap= 130"
# [1] "2 gene set: ImmuneSignature overlap= 135"
# 读取数据
scores_rs <- read.table("lihc_estimate_score.gct",skip = 2,header = T)
head(scores_rs)[1:3,1:6]
# NAME Description TCGA.FV.A495.01A TCGA.ED.A7PZ.01A TCGA.ED.A97K.01A TCGA.ED.A7PX.01A
# 1 StromalScore StromalScore -267.6186 -1691.1487 -631.36012 -297.48627
# 2 ImmuneScore ImmuneScore 747.3736 -903.0237 85.93637 -75.69938
# 3 ESTIMATEScore ESTIMATEScore 479.7550 -2594.1723 -545.42375 -373.18566

但这里输出结果里边是没有TumorPurity列(使用affymetrix平台的芯片数据会有)。如果没有就需要采用公式:Tumour purity = cos (0.6049872018+0.0001467884 × ESTIMATE score)去计算肿瘤纯度
rownames(scores_rs) <- scores_rs$NAME
scores_rs <- dplyr::select(scores_rs,-c(1:2))
TumorPurity = cos(0.6049872018+0.0001467884 * scores_rs[3,])
rownames(TumorPurity) <- "TumorPurity"
head(TumorPurity)
这里值得一提是基质分数,免疫分数,ESTIMATE分数和肿瘤纯度这四个概念:
1) 基质评分(Stromal Score)
高基质评分:通常表示肿瘤组织中存在较多的基质成分,如成纤维细胞和基质细胞。较高的基质评分在某些肿瘤类型中可能与较差的预后相关,因为这些细胞可以促进肿瘤的生长和转移,并可能阻碍免疫细胞的浸润和作用。
低基质评分:通常表示基质成分较少。在一些情况下,较低的基质评分可能与更好的预后相关,因为肿瘤微环境中缺乏支持肿瘤生长的基质细胞。
2) 免疫评分(Immune Score)
高免疫评分:通常表示肿瘤组织中有较多的免疫细胞浸润。高免疫评分通常被视为良好的预后标志,尤其是在某些肿瘤类型中,强烈的免疫反应可能有助于抑制肿瘤的生长和扩散。
低免疫评分:通常表示肿瘤组织中的免疫细胞较少,这可能与较差的预后相关,因为缺乏足够的免疫反应可能使肿瘤更容易逃避免疫监控。
3) ESTIMATE 评分(Estimate Score)
高 /低 ESTIMATE 评分:表示非肿瘤细胞的比例高或者低
4) 肿瘤纯度(Tumor Purity)
高/低肿瘤纯度:表示肿瘤细胞的比例高或者低
小结:
上述这6种免疫浸润方式,除了ESTIMATE需要采用log2之后的数据,其他RNA-seq的数据可以统一使用TPM(感觉这个最普适) ,芯片数据就不需要提了。最后得出的结果可以做很多可视化分析,比如多方法的整合热图。
此外,目前也有整合了多种免疫浸润分析R包可以使用,之后会重点介绍一下两个IOBR和immunedeconv这两个R包。
参考资料:
1、Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. 2019 Jul 15;35(14):i436-i445.
2、Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol. 2018:1711:243-259. doi: 10.1007/978-1-4939-7493-1_12 IF: NA NA NA.
3、xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017 Nov 15;18(1):220.
4、Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016 Oct 20;17(1):218.
5、TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017 Nov 1;77(21):e108-e110.
6、Deconvoluting tumor-infiltrating immune cells from RNA-seq data using quanTIseq. Methods Enzymol. 2020:636:261-285. doi: 10.1016/bs.mie.2019.05.056 IF: NA NA NA. Epub 2019 Jun 22.
7、Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017 Nov 13:6:e26476. doi: 10.7554/eLife.26476 IF: 6.4 Q1 B1.
8、ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and its Application in Cancer Immunotherapy. Adv Sci (Weinh). 2020 Feb 11;7(7):1902880.
9、Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013:4:2612. doi: 10.1038/ncomms3612 IF: 14.7 Q1 B1.
10、cibersort:https://github.com/Moonerss/CIBERSORT/blob/main/R/CIBERSORT.R
11、quanTIseq:https://icbi.i-med.ac.at/software/quantiseq/doc/index.html
12、EPIC:https://github.com/GfellerLab/EPIC/blob/master/vignettes/EPIC.Rmd
13、xCell:https://github.com/dviraran/xCell/tree/master/vignettes
14、MCPcounter:https://github.com/ebecht/MCPcounter
15、Estimate:https://rdrr.io/rforge/estimate/man/e00-estimate-package.html
16、被炸熟的虾: https://mp.weixin.qq.com/s/k7KbioTpnV_NiJW7JrrnaQ;
17、Bio小菜鸟: https://mp.weixin.qq.com/s/OqoxUyhD6_PsA_LbxD7Lcg
18、melonwd: https://mp.weixin.qq.com/s/Z8uGH5wFvvR4LFmTWGBY7w
19、生信益站: https://mp.weixin.qq.com/s/2X27dNODiBx8d-m2C9_y_A
20、生信图: https://mp.weixin.qq.com/s/Zdf4R5cgCiOlzc_o1xzDxg
21、生信菜鸟团: https://mp.weixin.qq.com/s/q7X8j4_os94jB3lbzMN_vA
22、生信技能树:
https://mp.weixin.qq.com/s/Z_zVPPfzr3DdCp0drpqgxA https://mp.weixin.qq.com/s/D-X3ZkyTSb6vyajo_aLDGg https://mp.weixin.qq.com/s/kFz56f0okY8TkSxKnJd3UQ https://mp.weixin.qq.com/s/JTD8ZmH2YYCIqcbs-97JzA https://mp.weixin.qq.com/s/UehaaJZgARryH7P25v9wNQ
致谢:感谢曾老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -