介绍
Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer提供了一种荟萃分析的框架,它主要基于常用的Wilcoxon rank-sum test和Blocked Wilcoxon rank-sum test 方法计算显著性,再使用分位数计算分组间的倍数变化,最后通过AUC判断物种的区分分组的能力。最后通过热图和森林图展示筛选到的在不同研究和荟萃分析均有差异的物种。
该框架可用于同类型的微生物荟萃分析。
加载R包
#| warning: false
#| message: false
library(tidyverse)
library(readr)
library(coin)
library(pROC)
library(RColorBrewer)
library(cowplot)
# rm(list = ls())
options(stringsAsFactors = F)
options(future.globals.maxSize = 1000 * 1024^2)
导入数据
数据下载百度云盘链接: https://pan.baidu.com/s/1VS6S8p5s20vwZ6FyILYoaQ
提取码: g4y3
-
物种表达谱数据
-
样本分组信息
#| warning: false
#| message: false
feat.all <- read.table("./data/meta-CRC-2019/feat_rel_crc.tsv",
sep='\t', header=TRUE,
stringsAsFactors = FALSE,
check.names = FALSE, quote='') %>%
as.matrix()
meta <- read_tsv('./data/meta-CRC-2019/meta_crc.tsv', show_col_types = FALSE)
- 其他参数(过滤和检验结果)
#| warning: false
#| message: false
alpha.meta <- 1e-05
alpha.single.study <- 0.005
mult.corr <- 'fdr'
pr.cutoff <- 0.05
log.n0 <- 1e-05
log.n0.func <- 1e-08
study.cols <- c('#2FBFBF', '#177254', '#F2CC30', '#74B347', '#8265CC')
数据预处理
-
提出研究名称
studies -
设置
block分组,用于后续检验
#| warning: false
#| message: false
studies <- meta %>%
dplyr::pull(Study) %>%
unique
# block for colonoscopy and study as well
meta <- meta %>%
dplyr::filter(!is.na(Sampling_rel_to_colonoscopy)) %>%
dplyr::mutate(block = ifelse(Study != 'CN-CRC', Study,
paste0(Study, '_', Sampling_rel_to_colonoscopy)))
feat.all <- feat.all[, meta$Sample_ID]
荟萃分析
荟萃分析采用了Wilcoxon rank-sum test和Blocked Wilcoxon rank-sum test 两种方法对单个研究和合并所有研究做显著性检验。本次需要计Foldchange(FC),单个研究的pvalue + 所有研究的pvalue(p.val)和单个研究和所有研究的AUC(aucs),以下是该代码的计算过程:
-
先使用Wilcoxon rank-sum test计算每个研究的每个物种在case/control之间的显著性检验结果;
-
再通过roc函数计算每个研究的每个物种在case/control之间的判别效果;
-
接着通过分位数quantile计算每个研究的每个物种在case/control之间的倍数变化;
-
然后通过Blocked Wilcoxon rank-sum test计算所有研究的荟萃差异检验结果;
-
最后计算所有研究的平均倍数变化作为整体倍数变化和通过roc函数计算每个物种在case/control之间的判别效果。
#| warning: false
#| message: false
p.val <- matrix(NA, nrow = nrow(feat.all), ncol = length(studies)+1,
dimnames = list(row.names(feat.all), c(studies, 'all')))
fc <- p.val
aucs.mat <- p.val
aucs.all <- vector('list', nrow(feat.all))
cat("Calculating effect size for every feature...\n")
pb <- txtProgressBar(max = nrow(feat.all), style = 3)
# caluclate wilcoxon test and effect size for each feature and study
for (f in row.names(feat.all)) {
# for each study
for (s in studies) {
x <- feat.all[f, meta %>% dplyr::filter(Study == s) %>%
dplyr::filter(Group=='CRC') %>% dplyr::pull(Sample_ID)]
y <- feat.all[f, meta %>% dplyr::filter(Study==s) %>%
dplyr::filter(Group=='CTR') %>% dplyr::pull(Sample_ID)]
# Wilcoxon: 对单个研究的单个物种检验
p.val[f, s] <- wilcox.test(x, y, exact=FALSE)$p.value
# AUC:评估每个物种区分分组的能力
aucs.all[[f]][[s]] <- c(roc(controls=y, cases=x,
direction='<', ci=TRUE, auc=TRUE)$ci)
aucs.mat[f, s] <- c(roc(controls=y, cases=x,
direction='<', ci=TRUE, auc=TRUE)$ci)[2]
# FC:使用10分位数计算每个物种的相对丰度再计算Foldchange结果
q.p <- quantile(log10(x+log.n0), probs = seq(.1, .9, .05))
q.n <- quantile(log10(y+log.n0), probs = seq(.1, .9, .05))
fc[f, s] <- sum(q.p - q.n)/length(q.p)
}
# calculate effect size for all studies combined
# Wilcoxon + blocking factor:计算所有研究混合在一起的检验结果
d <- data.frame(y = feat.all[f,],
x = meta$Group,
block = meta$block) %>%
dplyr::mutate(x = factor(x),
block = factor(block))
p.val[f, 'all'] <- coin::pvalue(wilcox_test(y ~ x | block, data = d))
# other metrics
x <- feat.all[f, meta %>% dplyr::filter(Group=='CRC') %>% dplyr::pull(Sample_ID)]
y <- feat.all[f, meta %>% dplyr::filter(Group=='CTR') %>% dplyr::pull(Sample_ID)]
# FC: 取所有样本的平均FC结果
fc[f, 'all'] <- mean(fc[f, studies])
# AUC:合并数据集每个物种区分不同分组样本的能力
aucs.mat[f, 'all'] <- c(roc(controls=y, cases=x,
direction='<', ci=TRUE, auc=TRUE)$ci)[2]
# progressbar
setTxtProgressBar(pb, (pb$getVal()+1))
}
cat('\n')
# multiple hypothesis correction
p.adj <- data.frame(apply(p.val, MARGIN=2, FUN=p.adjust, method=mult.corr),
check.names = FALSE)
查看结果
查看上述荟萃分析的结果
#| warning: false
#| message: false
head(p.adj)
head(aucs.mat)
head(fc)

画图
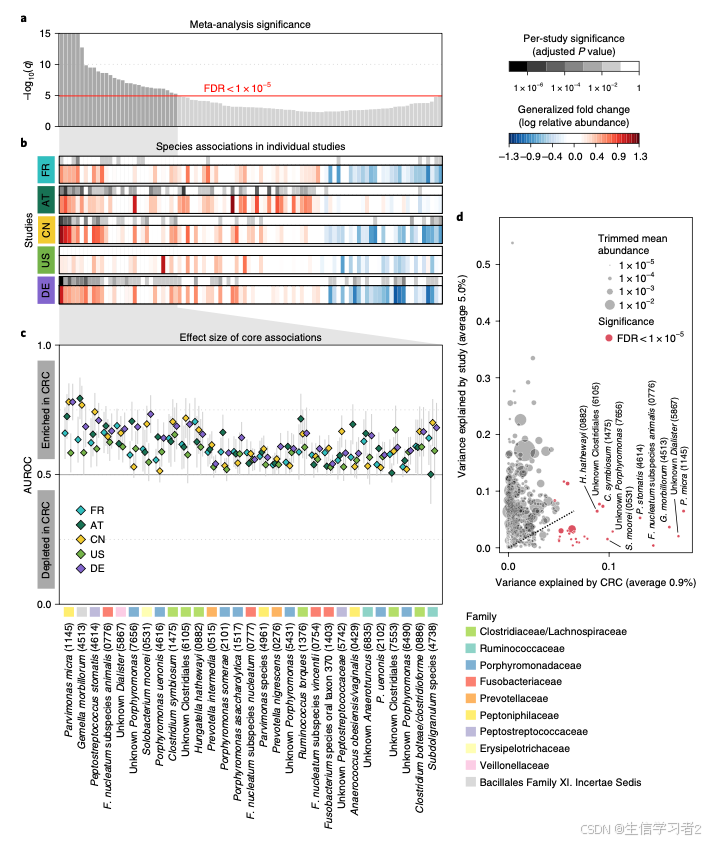
文章给出的图分成两部分,上部分是热图形式,下半部是森林图。
- 热图: 展示不同研究显著差异的物种
#| warning: false
#| message: false
species.heatmap <- rownames(p.adj)[which(p.adj$all < alpha.single.study)]
fc.sign <- sign(fc)
fc.sign[fc.sign == 0] <- 1
p.val.signed <- -log10(p.adj[species.heatmap,"all", drop=FALSE]) *
fc.sign[species.heatmap, 'all']
top.markers <- rownames(p.val.signed[is.infinite(p.val.signed$all) , , drop=FALSE])
p.val.signed[top.markers, 'all'] <- 100 + aucs.mat[top.markers, 'all']
species.heatmap.orderd <- rownames(p.val.signed[order(p.val.signed$all), , drop=FALSE])
# take only those
fc.mat.plot <- fc[species.heatmap.orderd, ] %>% as.data.frame()
p.vals.plot <- p.adj[species.heatmap.orderd, ]
# ##############################################################################
# prepare plotting
# colorscheme for fc heatmap
mx <- max(abs(range(fc.mat.plot, na.rm=TRUE)))
mx <- ifelse(round(mx, digits = 1) < mx,
round(mx, digits = 1) + 0.1,
round(mx, digits = 1))
brs <- seq(-mx, mx, by=0.05)
num.col.steps <- length(brs) - 1
n <- floor(0.45*num.col.steps)
col.hm <- c(rev(colorRampPalette(brewer.pal(9, 'Blues'))(n)),
rep('#FFFFFF', num.col.steps-2*n),
colorRampPalette(brewer.pal(9, 'Reds'))(n))
# color scheme for pval heatmap
alpha.breaks <- c(1e-06, 1e-05, 1e-04, 1e-03, 1e-02, 1e-01)
p.vals.bin <- data.frame(apply(p.vals.plot, 2, FUN=.bincode,
breaks = c(0, alpha.breaks, 1),
include.lowest = TRUE),
check.names = FALSE)
p.val.greys <- c(paste0('grey',
round(seq(from=10, to=80,
length.out = length(alpha.breaks)))),
'white')
names(p.val.greys) <- as.character(1:7)
# function to plot both into a grid
plot.single.study.heatmap <- function(x) {
# x = "FR-CRC"
df.plot <- tibble(species = factor(rownames(p.vals.plot),
levels = rev(rownames(p.vals.plot))),
p.vals = as.factor(p.vals.bin[[x]]),
fc = fc.mat.plot[[x]])
g1 <- df.plot %>%
ggplot(aes(x = species, y = 1, fill = fc)) +
geom_tile() + theme_minimal() +
theme(axis.text = element_blank(),
axis.ticks = element_blank(),
axis.title = element_blank(),
panel.grid = element_blank(),
panel.background = element_rect(fill=NULL, colour='black'),
plot.margin = unit(c(0, 0, 0, 0), 'cm')) +
scale_y_continuous(expand = c(0, 0)) +
scale_fill_gradientn(colours=col.hm, limits=c(-mx, mx), guide=FALSE)
g2 <- df.plot %>%
ggplot(aes(x=species, y=1, fill=p.vals)) +
geom_tile() + theme_minimal() +
theme(axis.text = element_blank(),
axis.ticks = element_blank(),
axis.title = element_blank(),
panel.grid = element_blank(),
panel.background = element_rect(fill=NULL, colour='black'),
plot.margin = unit(c(0, 0, 0, 0), 'cm')) +
scale_y_continuous(expand = c(0, 0)) +
scale_fill_manual(values=p.val.greys, na.value='white', guide=FALSE)
g.return <- plot_grid(g2, g1, ncol = 1, rel_heights = c(0.25, 0.75))
return(g.return)
}
# ##############################################################################
# plot
# p.value histogram
g1 <- tibble(species = factor(rownames(p.vals.plot),
levels = rev(rownames(p.vals.plot))),
p.vals = -log10(p.vals.plot$all),
colour = p.vals > 5) %>%
ggplot(aes(x = species, y = p.vals, fill = colour)) +
geom_bar(stat = 'identity') +
theme_classic() +
xlab('Gut microbial species') +
ylab('-log10(q-value)') +
theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.ticks.x = element_blank(),
axis.text.x = element_blank(),
panel.background = element_rect(fill = NULL, color = 'black')) +
scale_y_continuous(limits = c(0, 15), expand = c(0, 0)) +
scale_x_discrete(position = 'top') +
scale_fill_manual(values = c('lightgrey', 'darkgrey'), guide = FALSE)
g.lst <- lapply(studies, plot.single.study.heatmap)
pl1 <- plot_grid(g1, g.lst[[1]], g.lst[[2]],
g.lst[[3]], g.lst[[4]],g.lst[[5]],
ncol = 1, align = 'v',
rel_heights = c(0.3, rep(0.12, 5)))
pl1

结果:差异物种在不同研究和整合数据的倍数变化和显著性结果
-
最上图是物种在整合数据的显著性结果(adjustedPvalue);
-
接下来的热图是物种在单个研究的显著性结果(上半部)和倍数变化(下半部:红色是富集在CRC,蓝色是CTRL);
-
森林图: 不同物种在每个研究区分case/control的能力 (通过alpha.meta更严格筛选)
#| warning: false
#| message: false
# select and order
marker.set <- rownames(p.val.signed)[
abs(p.val.signed$all) > -log10(alpha.meta)]
p.val.signed.red <- p.val.signed[marker.set, ,drop=FALSE]
marker.set.orderd <- rev(rownames(p.val.signed.red[order(p.val.signed.red$all),,
drop=FALSE]))
# extract those from the auc list
df.plot <- tibble()
for (i in marker.set.orderd){
for (s in studies){
temp <- aucs.all[[i]][[s]]
df.plot <- bind_rows(df.plot, tibble(
species=i, study=s,
low=temp[1], auc=temp[2], high=temp[3]
))
}
}
df.plot <- df.plot %>%
dplyr::mutate(species = factor(species, levels = marker.set.orderd)) %>%
dplyr::mutate(study = factor(study, levels = studies))
# plot everything
pl2 <- df.plot %>%
ggplot(aes(x = study, y = auc)) +
geom_linerange(aes(ymin = low, ymax = high), color = 'lightgrey') +
geom_point(pch = 23, aes(fill = study)) +
facet_grid(~species, scales = 'free_x', space = 'free') +
theme_minimal() +
scale_y_continuous(limits=c(0, 1)) +
theme(panel.grid.major.x = element_blank(),
axis.ticks.x = element_blank(),
axis.text.x = element_blank(),
strip.text = element_text(angle=90, hjust=0)) +
scale_fill_manual(values = study.cols,
guide = FALSE) +
ylab('AUROC') + xlab('Gut microbial species')
pl2

- 合并图: 最后文章呈现的图是经过修改的
#| warning: false
#| message: false
cowplot::plot_grid(pl1, pl2, ncol = 1)
总结
在进行荟萃分析时,本研究采用了一种特定的统计方法——Blocked Wilcoxon rank-sum test,以评估和整合不同研究中的case/control物种的显著性结果。该方法特别适用于处理微生物数据这类稀疏性数据集,因为它能够在计算两组之间的倍数变化时有效避免零值过多的问题。通过使用分位数方法,研究者能够更准确地估计和比较不同组之间的差异,从而提高了分析结果的可靠性和有效性。
对于类似类型的研究,研究者可以采用与本研究相似的分析框架进行荟萃分析。这包括以下几个关键步骤:
- 数据的收集与整理:确保收集到的数据是高质量的,并且适合进行荟萃分析。
- 选择合适的统计方法:根据数据的特点选择合适的统计检验方法,如Blocked Wilcoxon rank-sum test,以确保分析的准确性。
- 数据处理:对于稀疏数据,采用分位数方法来处理零值过多的问题,以提高分析的稳健性。
- 结果的整合与解释:将不同研究的结果进行整合,并采用适当的统计方法来评估整体的显著性。
通过遵循这样的框架,研究者可以对类似主题的研究进行系统性地分析和比较,从而为该领域的研究提供更深入的见解。