—DOI: 10.1038/s41592-024-02325-3
Systematic comparison of sequencing-based spatial transcriptomic methods
学习了一下空间转录组技术怎么做benchmark,从多个的角度去考虑目前技术的性能,受益良多。但该研究缺少对10X Visium HD的测评,可以期待一下后续的文章。
留意更多内容,欢迎关注微信公众号:组学之心
主要研究团队和单位
:::: column
::: column-left
刘晓东–西湖大学生命科学学院
:::
::: column-right
田鲁亦–广州实验室

:::
::::
文章简介
1.背景
基于测序的空间转录组学(sST)促进了空间基因表达测量的发展,sST 能够在保留组织空间信息的同时进行全面的转录组学分析。尽管基于成像的空间转录组学已经开展了基准测试,但sST 仍处于早期发展阶段,尚未进行系统的基准研究。sST 技术在使用空间 DNA 条形码等方面具有共同特征,其在空间分辨率和条形码阵列制备方面存在显著差异,这使得方法选择和评估标准的制定变得复杂。目前缺乏对不同平台的全面基准测试,技术和数据集的差异性使标准化评估指标的制定变得困难。
2.研究目的
研究系统比较了11种sST方法(包括 10X Genomics Visium(基于 poly-A 和基于探针的两种方法)、DynaSpatial、HDST、BMKMANU S1000、Slide-seq V2、Curio Seeker(Slide-seq 的商业版本)、Slide-tag、Stereo-seq、PIXEL-seq、Salus 和 DBiT-seq),通过使用小鼠胚胎眼、小鼠大脑海马区和小鼠嗅球等参考组织生成跨平台基准数据集 cadasSTre。研究评估了每种技术在空间分辨率、捕获效率和分子扩散方面的性能,并更新了scPipe以预处理和下采样 sST 数据,减少变异性并促进技术整合。分析表明,不同 sST 技术在下游应用(如聚类、区域注释和细胞间通讯)中的表现各异,特别是基因检测偏差。
研究结果如下:
1.基准参照组织和实验设计
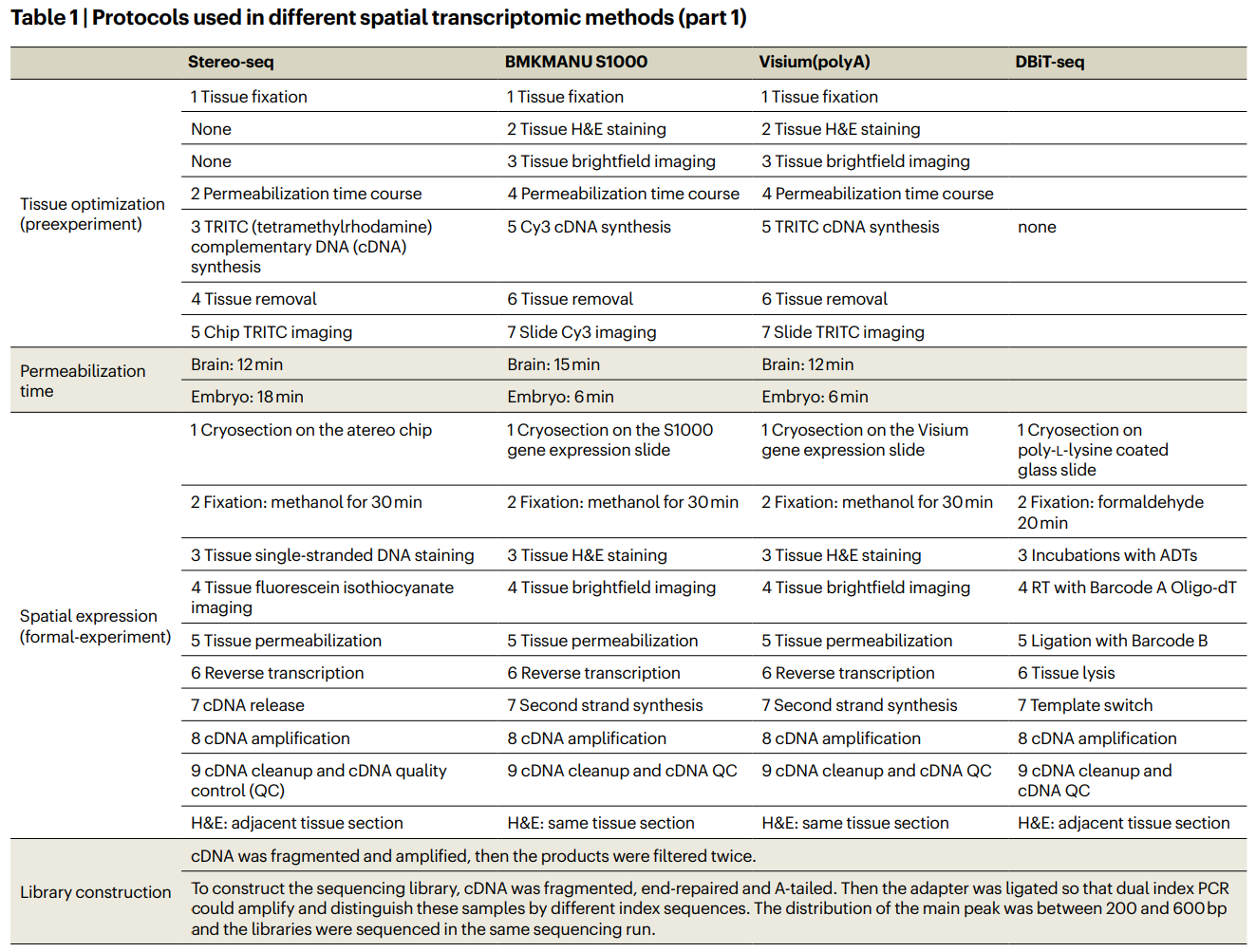
不同空间转录组学方法中使用的方案如下(样本预处理步骤和测序步骤):

样品、测序技术和数据分析流程如下:
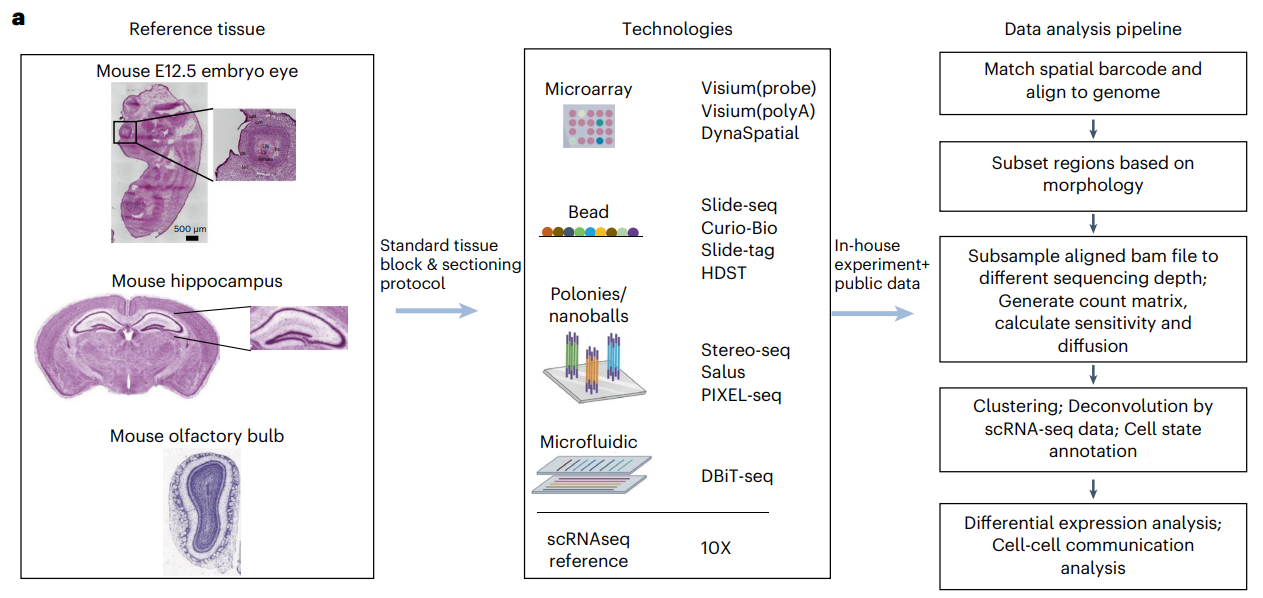
成年小鼠大脑、E12.5 小鼠胚胎和成年小鼠嗅球作为参考组织,因为它们具有相对明确的形态特征。这些组织具有已知的形态模式和异质性表达谱,作为sST基准研究的理想参考样品。
测序技术分为五种:
- Microarray:Visium (probe)、Visium (polyA)、DynaSpatial
- Bead:Slide-seq、Curio-Bio、、Slide-tag、HDST
- Polonies/nanoballs:Stereo-seq、Salus、PIXEL-seq
- Microfluidic:DBiT-seq
- scRNAseq reference:10X
数据分析流程主要包括以下步骤:
- 1.匹配空间条形码并对齐到基因组
- 2.基于形态学选择区域
- 3.将对齐的 bam 文件下采样到不同的测序深度,生成计数矩阵,计算灵敏度和扩散
- 4.聚类;通过 scRNA-seq 数据去卷积;细胞状态注释
- 5.差异表达分析;细胞-细胞通信分析
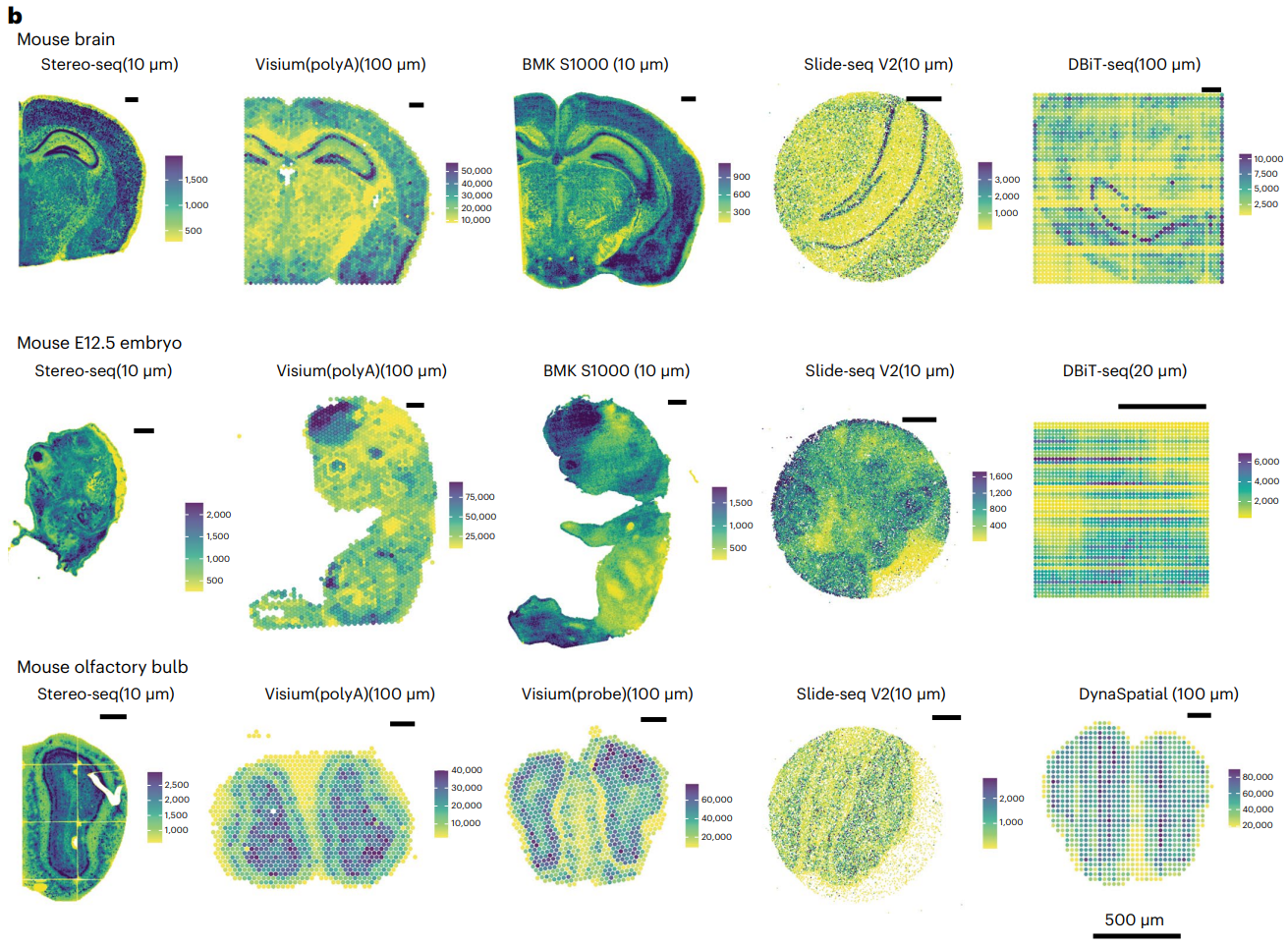
图中很好的显示了不同测序技术的分辨率大小,组织捕获的范围以及测序深度。Stereo-seq、BMKMANU S1000 和 Salus 的测序位点中心之间的距离小于 10 μm,测序位点被分成 10 μm 大小的点以便于可视化。Stereo-seq、Visium、BMKMANU S1000 和 Salus 成功捕获了几乎整个右脑和整个 E12.5 胚胎。

在这些方法中,Stereo-seq 表现出最高的捕获能力。其常规测序芯片大小为 1 厘米,最大尺寸可达 13.2 厘米。相比之下,Slide-seq V2 由于捕获尺寸有限,只能捕获部分组织。而DBiT-seq捕获尺寸会根据微流体通道的宽度而变化。随后,研究选择性地保留已知形态区域内的读数,然后进行降采样,以解决测序深度和测序成本变化问题。最后,生成具有降采样数据和完整数据的count矩阵,用于灵敏度和扩散计算,然后进行细胞状态注释、标记基因检测和细胞间通讯分析。
降采样是将原始测序数据中的读数减少到一个目标数量,以模拟不同测序深度下的数据表现。这么做可以将所有方法的数据降采样到相同的读数数量,可以消除因测序深度不同而导致的比较不公平的问题,此外,测序深度直接影响测序成本。将不同方法的数据降采样到相同的读数数量,使得每种方法的成本相对等效,从而使得基于成本的比较更有意义。
2.分子捕获效率测评
分子捕获效率以两种方式评估。在选定的区域中,
- ①使用该区域的total counts;
- ②对数据进行下采样,得到下采样数据,以便不同的样本具有相同数量的测序counts。
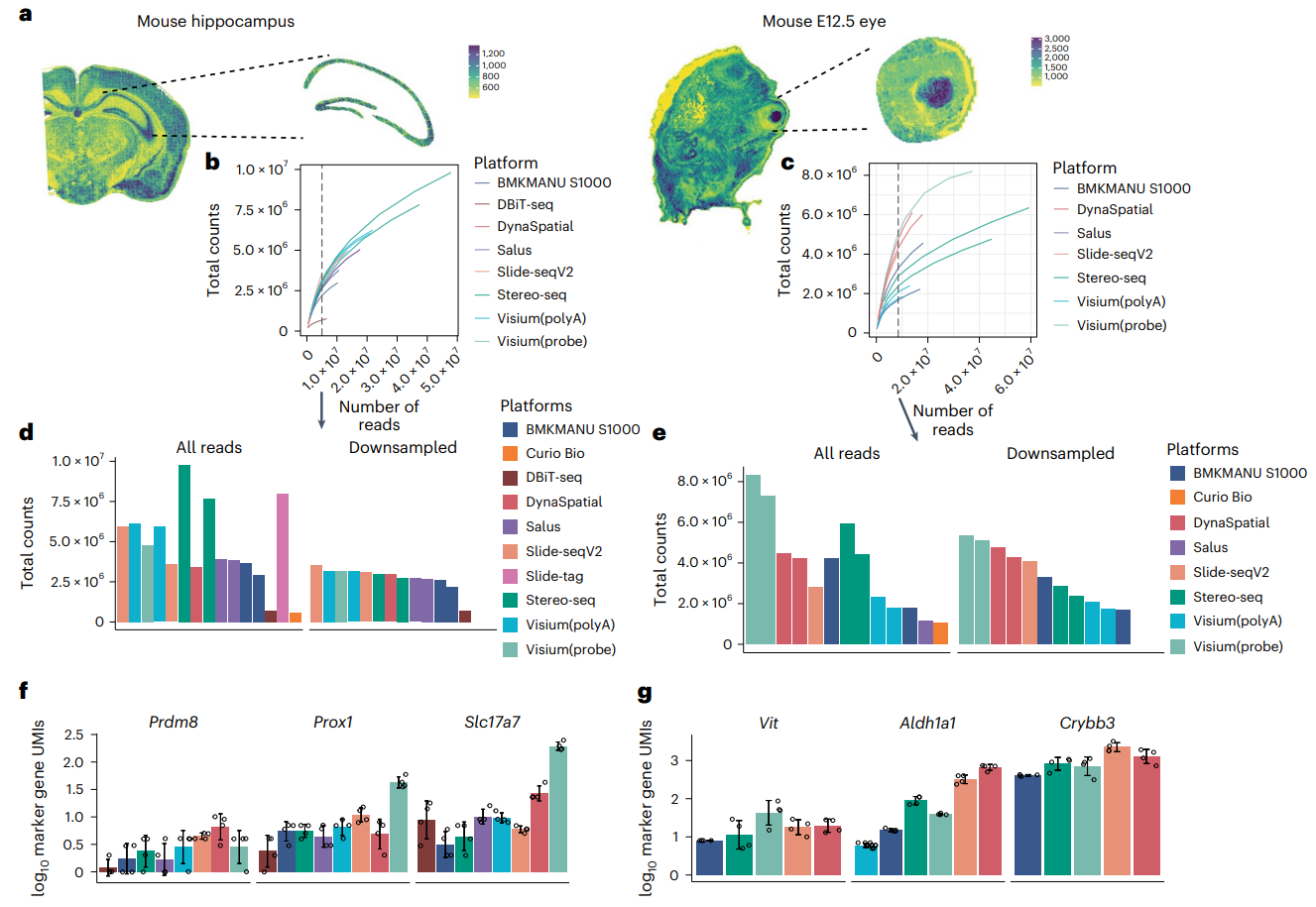
(a)通过手动划定边界从成年小鼠大脑和 E12.5 小鼠胚胎中获取了海马和眼组织。
(b-c)根据下采样结果,所有测序run(范围从 3 亿个reads(Visium)到 40 亿个reads(Stereo-seq))均未达到饱和。这一观察结果表明,sST数据需要更多的reads才能达到最佳性能,并且可能会提高灵敏度。
(d-e 左)与其他平台相比,Stereo-seq 在小鼠海马和眼睛中对同一区域具有更多的测序reads,因此当使用all reads时,总计数相对较高。尽管 Visium(探针)的测序深度几乎是Stereo-seq的一半,但它在小鼠眼中呈现出最高的all reads。这可能是因为基于探针的方法具有更好的读取效率,以及使用探针时过度量化的唯一分子标识符 (UMI) 计数的潜在影响。
(d-e 右)使用下采样数据时,Slide-seq V2 数据在小鼠眼中表现出比其他平台更高的灵敏度,而在海马中,基于探针的 Visium、DynaSpatial,然后是 Slide-seq V2 表现出更高的灵敏度。
(f-g)使用下采样数据测量了已知在特定区域表达的标记基因的 RNA 含量。海马组织中,Visium(probe)、Slide-seq V2 和 DynaSpatial 在海马中表现出最高的敏感性。小鼠眼组织中,类似地,Visium(probe)、DynaSpatial、Slide-seq V2 表现出最高的灵敏度。

(h)比较Visium(polyA)和Stereo-seq之间检测到的基因的总UMI计数。每个点代表一个基因,以黑色显示。红色基因是在Stereo-seq中在表达量很高的,但在Visium(polyA)中表达量很低。
(i)热图显示了E12.5 小鼠眼睛组织的基因表达,这些基因没有被 Visium(polyA) 捕获,但被 Stereo-seq 捕获。
Visium(polyA)测序技术对某些蛋白质编码基因存在系统性偏差,使得这些基因在Visium(polyA)中的检测表达水平显著低于在Stereo-seq中的水平。这种偏差与预处理流程、基因注释、GC含量和基因长度无关,主要是由于测序技术本身对特定基因的检测敏感性不同所致。
3.分子横向扩散测评–mRNA检测的空间精度
作者使用了两种分析方法来测量分子横向扩散(all reads生成的count数据):
- ①在选定区域内绘制特定基因的密度分布图。这种图展示了基因表达在空间上的分布情况,从而可以直观地看到基因表达的扩散模式。
- ②量化选定区域内强度半峰左宽(LWHM)之间的距离。LWHM是指强度分布曲线在一半最大值处的宽度,通过测量这个宽度可以定量地描述基因表达在空间上的扩散程度。

(a)Biomarker包括小鼠嗅球中的Slc17a7(左)、小鼠脑中的Ptgds(中)和E12.5眼中的Pmel(右)。其中黑框表示用于扩散计算的选定区域。
(b-d)(左)基于强度图和 LWHM 测量的观察结果显示,Stereo-seq 在嗅球中显著横向扩散 Slc17a7,而Slide-seq V1.5 和 PIXEL-seq 对这种扩散的控制相对较好; (中)Ptgds的表达图及其密度图以及 LWHM 测量值显示Stereo-seq数据集中存在严重的横向扩,相比之下,Slide-seq V2 和 BMKMANU S1000 对此类横向扩散问题表现出更好的控制;(右)检测Pmel标记基因时,Stereo-seq 表现出对横向扩散的最佳控制,其次是 Slide-seq V2。
由此可见,组织类型对扩散过程有相当大的影响。另一个因素是通透时间,它对扩散模式和 mRNA 捕获有很大影响。

而在分辨率较低的方法中,Visium(polyA) 更清晰地保留了嗅球各层中 Slc17a7 表达的两个峰,而 DynaSpatial 几乎没有提供分离的表达峰。

此外,大脑组织检测Ptgds基因时,发现下采样无法解决Stereo-seq数据的横向扩散问题。
4.聚类和细胞注释结果测评
4.1 聚类结果比较 & 降采样对聚类的影响

比较了三种不同的聚类方法:Seurat(专门考虑转录组谱)、DR.SC31 和 PRECAST32(结合空间信息和基因表达数据)。Seurat 在检测预期细胞亚群方面始终表现出比其他两种方法更稳健和稳定的性能,因此后续分析主要关注 Seurat 生成的结果。
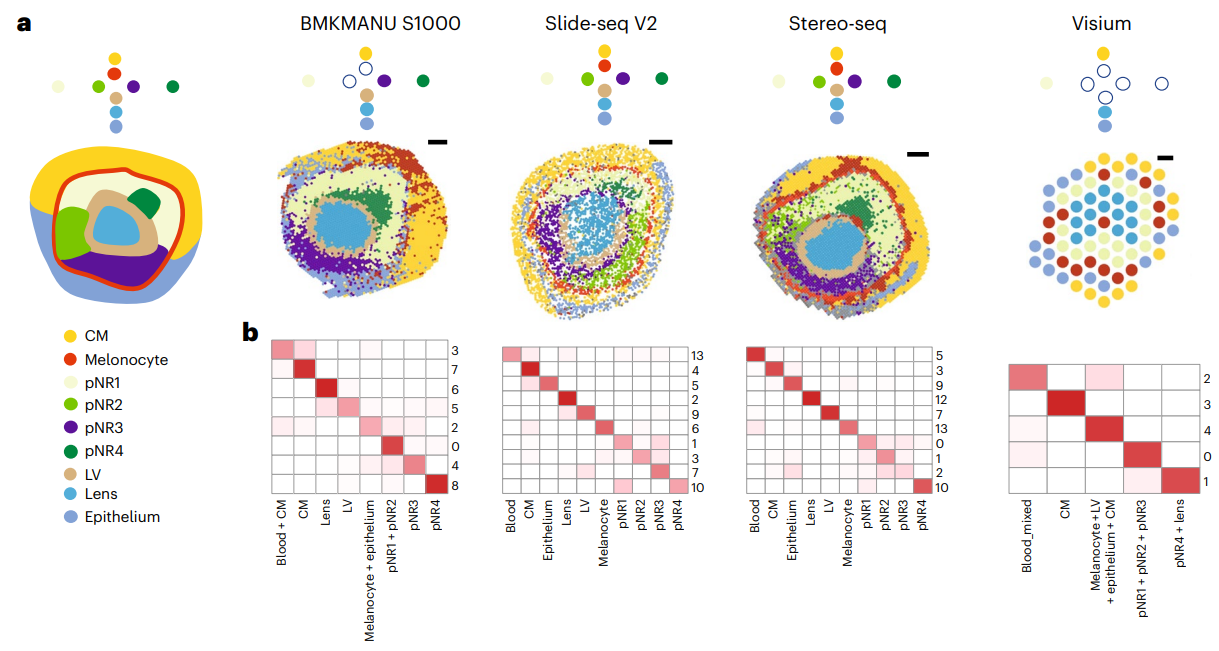
(a)最左边的图是E12.5 小鼠眼细胞亚群的参考。Slide-seq V2 和 Stereo-seq 数据提供了良好的spot分离,可进行全面的子集注释,成功捕获所有预期的子集。BMKMANU S1000 数据较差,这种困难可能源于这技术有明显的横向扩散,这使得仅依赖表达谱的聚类方法难以保留这种特定的细胞类型。而分辨率为 100 μm 的数据(包括 Visium 和 DynaSpatial)在检测预期的细胞亚群方面面临一定的限制。
(b)测序深度影响空间转录组数据的all reads。因此需要对降采样数据的聚类结果进行探索,涉及两个关键方面:
- (1)评估了降采样数据与完整数据之间的对应关系。
- (2)以all reads作为参考,以不同比例生成的降采样数据为基础,计算了聚类纯度(ECP)和准确度(ECA)的熵度量。
研究发现降采样数据能够检测到全数据识别的几乎所有细胞亚群。而评估不同比例值的 ECP 和 ECA 时,观察到相对较高的值,表明存在明显的不一致性。
这种不一致可能是因为:虽然大多数细胞亚群有效地形成了不同的簇,但一部分细胞聚集成不同的簇,尤其是在来自不同神经元视网膜细胞亚群的细胞之间。这种效应在晶状体和晶状体囊泡等群体之间的亚群以及四个神经元视网膜亚群中尤为明显,这些亚群的表达谱更为相似。
4.2 sST 数据与 snRNA-seq 数据之间的比较
附图21:

附图22:
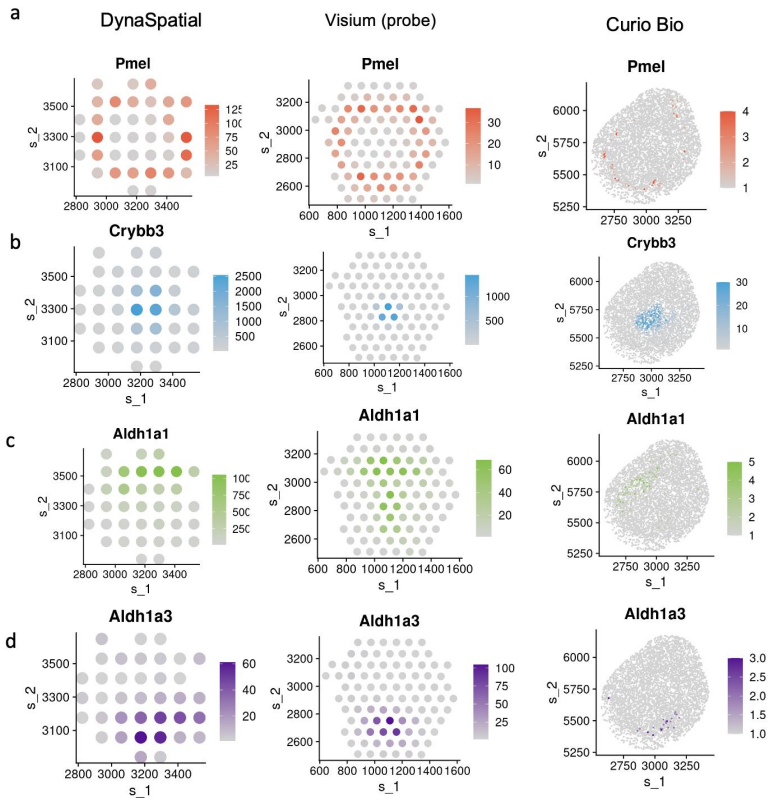
研究在所有 sST 数据集中一致观察到 Pmel、Crybb3、Atoh7、Enfa5、Aldh1a1 和 Aldh1a3 的良好模式表达。之所以选择这些基因,是因为它们可作为特定细胞类型的标记,例如黑素细胞、晶状体和假定神经视网膜 (pNR)2 和 pNR3。眼部区域snRNA-seq 数据显示只有有限数量的细胞表达上述基因(附图21e)。Crybb3 表达值相对低于预期,Crybb3 在 Visium(polyA) 中捕获得很少,表明基于 polyA 的 Visium 技术可能存在捕获偏差。
附图23:

而某些区域的绝对位置并不完全相同,但它们的相对位置保持一致,例如 Aldh1a3 位于视网膜层的喙区域,而 Aldh1a1 则朝向尾部区域。
附图24:
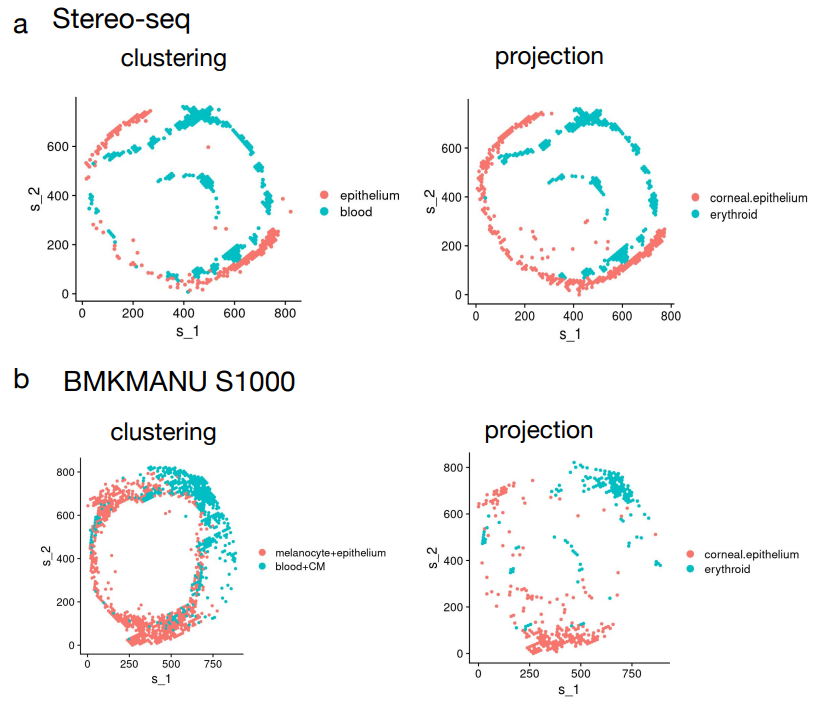
虽然 snRNA-seq 数据可能无法像 sST 方法那样捕获眼部区域中的那么多细胞,但它可作为注释 sST 数据的有用参考数据集。如附图24所示,使用 Seurat 将 snRNA-seq 数据与 sST 数据整合有助于注释 sST 数据。它改进了立体测序数据中上皮细胞的注释,这一直相对具有挑战性,因为存在具有混合表达谱的未知细胞簇。使用 snRNA-seq 上皮细胞投影可以更好地解析该簇。此外,投影有助于分离 BMKMANU S1000 数据中的黑色素细胞和上皮细胞。
附图25:

sST 技术易受血液污染,血液污染通常在组织制备和切片过程中引入,很难避免。以 Hba-a1 基因作为评估指标,发现Visium、DynaSpatial 和 BMKMANU S1000 受到血液污染的影响显著,所有 Visium 和 DynaSpatial 测序位点以及 70% 的 BMKMANU S1000 测序位点均表达 Hba-a1。相比之下,与 snRNA-seq 相比,Stereo-seq 数据的血液污染程度相对相似,而 Slide-seq V2 的血液污染程度最低。
5.标记基因检测结果测评
Wilcoxon 秩和检验在识别标记基因时的有效性和稳健性都很好,因此研究在Seurat 中使用此检验来查找簇之间的标记基因。
通过对top标记基因的分析,揭示了技术选择中的特定偏差。例如,Pax6 是一种在神经谱系中,尤其是在视网膜中,起主要调节作用的转录因子。
不同技术对 Pax6 的表达表现出差异:
- Stereo-seq 数据仅在 pNR3 簇中突出了 Pax6。
- Slide-seq V2 和 BMKMANU S1000 数据则显示 Pax6 在整个神经视网膜(pNR1-4)中均有表达。


(上c,下a)对下采样数据的聚类结果的分析表明,即使测序读数较少,一般细胞亚群仍然可以充分保留。然而那些有相似基因表达的细胞亚群就很难清楚地分离。为了进一步研究下采样的影响,将下采样数据中识别的标记基因与完整数据集中的标记基因进行了比较。研究选择了两对细胞亚群来比较具有相对相似表达谱和更明显表达谱的细胞亚群的检测性能。(1)表现出更高相似性的 pNR4 和 pNR1,以及(2)相似性较低的晶状体和黑素细胞。
(上d,下b)在两对比较中,随着读取次数的增加,标记基因的数量也随之增加。而且随着测序的深入,标记基因的增加更为明显。在具有相对不同表达谱的细胞亚群之间的比较中,sST 方法中标记基因检测性能的排名与图 2d 中所示的结果一致。特别是,Slide-seq V2 表现出更高的灵敏度。
(e)左下角的条形图显示了每个平台(Stereo-seq, BMK, Slide-seq V2)中标记基因的总数(Set size)。右侧的柱状图展示了不同平台之间标记基因的交集大小(Intersection size)。颜色区分了不同的组合:紫色部分表示标记基因在三个平台中共享的情况。粉色部分表示标记基因在两个平台中共享的情况。跨三个技术平台共享的标记基因数量较少(11个)。标记基因在两个平台中共享的数量稍多(16个在BMK和Slide-seq V2;3个在Stereo-seq和BMK)。在某个单一平台上独特的标记基因数量最多(26个在Slide-seq V2;21个在BMK;24个在Stereo-seq)。每个平台的独特标记基因数量比跨技术共享的标记基因更多,这表明不同技术平台在标记基因的识别上存在较大的差异。这种差异可能是由于每个平台的技术特点、测序深度、数据处理方法等因素导致。
6.总结
6.1 评估空间转录组学方法比评估单细胞RNA测序(scRNA-seq)方法更具挑战性
- ①设计参考组织的难度:空间转录组学参考组织的设计更为复杂。如果使用真实组织,细胞类型和基因表达模式明确,但位置和基本事实变得模糊,并受限于对参考组织的理解。而scRNA-seq可以使用细胞系混合物或外周血单核细胞样本获得一致输入。
- ②测量单位的差异:不同方法的测量单位不同。例如,Visium(基于polyA和基于探针)和DynaSpatial方法的斑点直径大于50μm,类似于mini-bulk RNA-seq。Stereo-seq等方法的斑点大小为亚微米,比单个细胞小得多。
6.2 应对挑战的基准研究设计
- ①选择参考组织的标准:参考组织应来自广泛使用的模型生物,便于大多数研究机构获取,而且应具有稳定的细胞类型模式和特异性标记基因表达。参考区域应具有清晰的形态,易于在切片中找到。
- ②使用多个基准指标和工作流程比较同一组织区域的不同方法,包括all reads和降采样数据的比较。此外,降采样用来减轻测序深度和成本变化的影响,但由于不同方法所需的读取次数不同,分析中还使用了all reads作为补充结果。
6.3 基准测试平台和结果
cadasSTre:在genographix.com上生成了cadasSTre,这是一个用于sST基准测试的跨平台数据集。用于系统评估35项实验中的11种sST方法,比较数据的各个方面,从基本指标到下游分析,包括敏感性、扩散性、聚类性和标记基因检测.
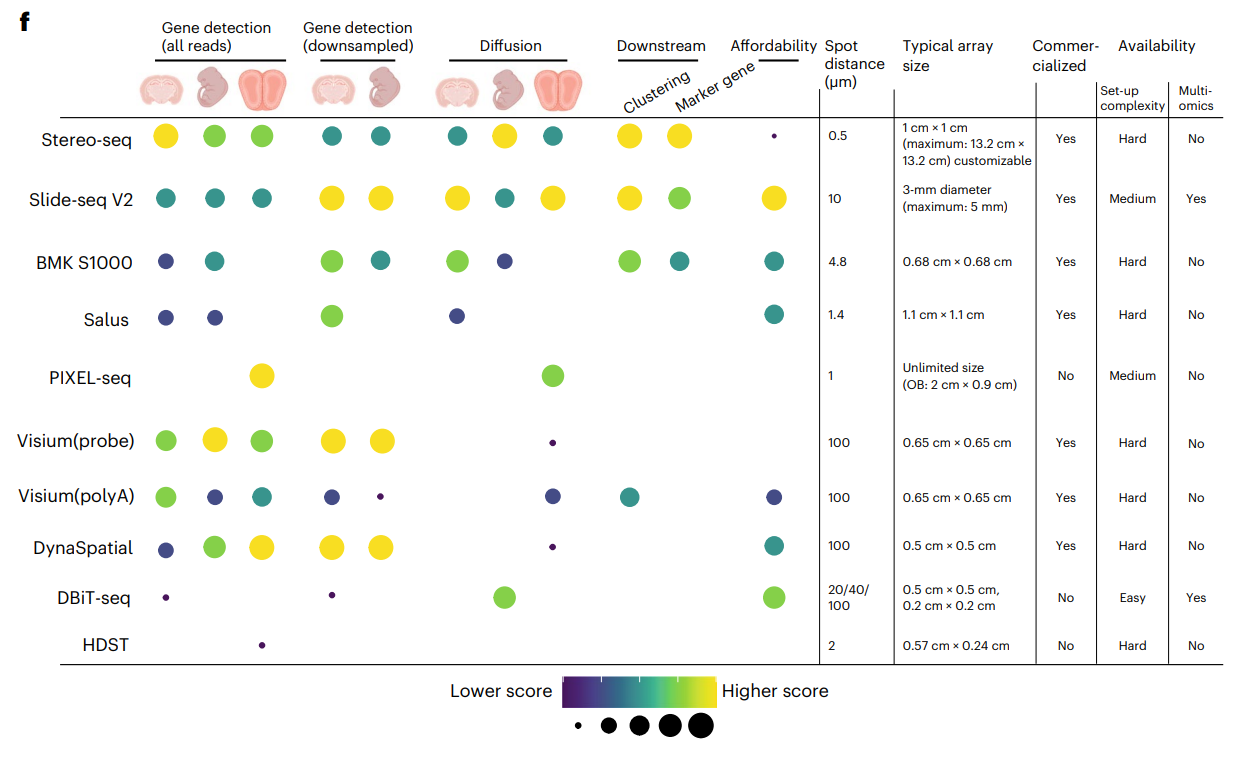
评估结果:
- 空间转录组学需要更多的测序才能达到饱和,生成的数据远低于饱和水平。
- Stereo-seq、Slide-tag、Visium(探针)在原始测序深度下显示出更好的捕获效率,而Slide-seq V2、Visium(探针)、DynaSpatial在标准化测序深度下表现更好。
- 基于polyA的Visium平台存在意外的基因捕获偏差,其他技术一致捕获的标记基因未在Visium(polyA)数据中出现。考虑到Visium是最广泛使用的商业平台,进一步验证其在其他组织上的基因捕获偏差非常重要。