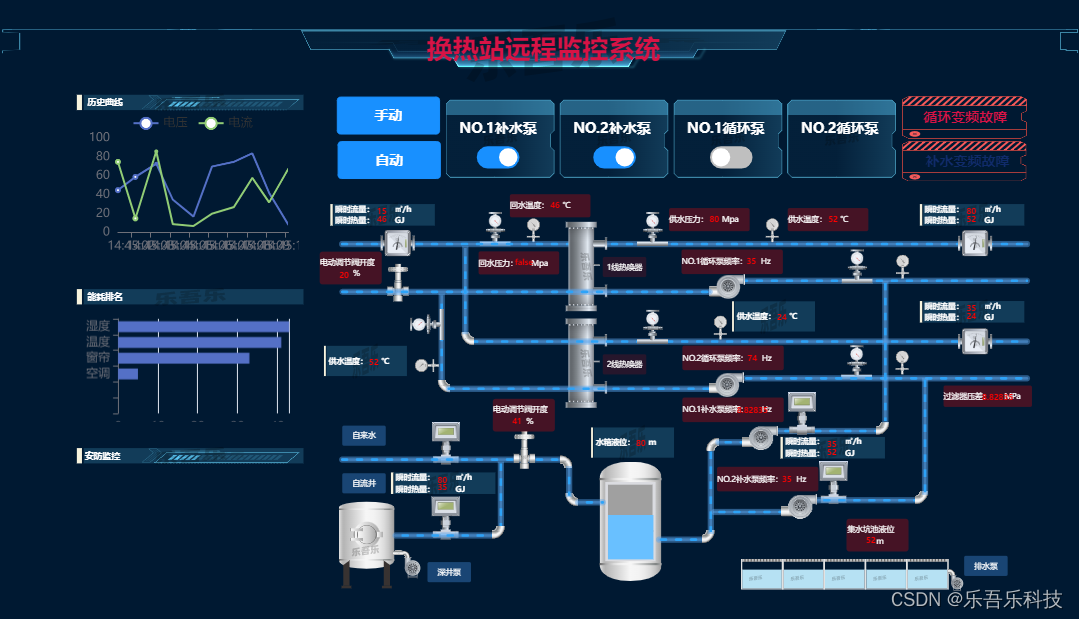
--------各项指标的计算--------
vcf=xxx.vcf.gz
out=xxx # 计算完的文件会自动生成文件后缀
# 1.计算每个个体的SNP的平均测序深度
vcftools --gzvcf $vcf --depth --out $out
# 2.计算每个SNP位点的测序深度
vcftools --gzvcf $vcf --site-mean-depth --out $out
# 3.计算每个SNP位点的质量
vcftools --gzvcf $vcf --site-quality --out $out
# 4.计算每个个体包含缺失位点的比例
vcftools --gzvcf $vcf --missing-indv --out $out
# 5.计算每个SNP位点的在所有个体缺失位点的比例
vcftools --gzvcf $vcf --missing-site --out $out
# 6.计算每个个体的杂合度和近交系数(判断是否有离群样本)
vcftools --gzvcf $vcf --het --out $out
引用参考自:四、重测序数据分析之变异识别与过滤
--------预览生成的各个文件的结构--------
此步可以省略,后续在R中导入后也可以看到。

图1 每个个体的SNP的平均测序深度
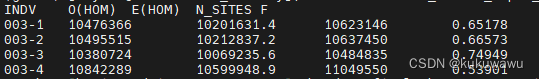
图2 每个个体的杂合度和近交系数
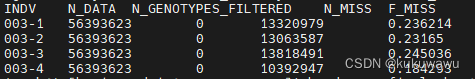
图3 每个个体包含缺失位点的比例
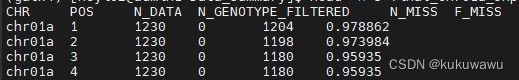
图4 每个SNP位点的在所有个体缺失位点的比例

图5 每个SNP位点的测序深度
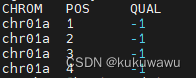
图6 每个SNP位点的质量
--------R ggplot绘制密度图--------
请先确保R软件和ggplot包已安装。
# 1. 导入ggplot2绘图包
library(ggplot2)
# 2. 导入文件
raw_idepth <- read.table("H:/filterbasis/final_chr01a_snp01_depth.idepth", header = TRUE)
# 读取文件时,不指定 sep 参数,让它自动检测分隔符;
# 指定header参数为TRUE,自动识别第一列为列名;
# 导入后,检查一下行列是否被正确识别。
# 3. 根据已知的列名,将该列进行数值化
raw_idepth$MEAN_DEPTH <- as.numeric(raw_idepth$MEAN_DEPTH)
# 如果不知道列名,可以用以下命令查看
print(colnames(raw_idepth))
# 4. 绘图
ggplot(raw_idepth, aes(x = MEAN_DEPTH)) + # 指定文件和列
geom_density(fill = "red", alpha = 0.5) + # 指定填充颜色和透明度
labs(title = "个体平均测序深度", x = "MEAN_DEPTH", y = "Density") # 设置表头和坐标轴绘制完成后如图7所示,可根据这个图来确定大部分数据所在范围,从而选择后续过滤最合适的参数。