首先,你需要确保你已经安装了R和RStudio(如果你想用RStudio的话)。然后,你可以按照以下步骤进行操作:
- 加载数据:首先,你需要加载你的表型数据。如果你的数据是以CSV、Excel等格式保存的,你可以使用
read.csv()或read.xlsx()等函数加载数据。
# 举例,假设你的数据是以CSV格式保存的 data <- read.csv("path/to/your/data.csv")
质量检查:一旦数据加载完成,你可以使用以下方法来进行质量检查:
head(data):查看数据的前几行,确保数据加载正确。
str(data):查看数据的结构,包括变量的类型和数量。
summary(data):获取数据的摘要统计信息,如均值、中位数、最小值、最大值等。
any(is.na(data)):检查是否有缺失值。
colSums(is.na(data)):查看每个变量中的缺失值数量。-
统计分析:根据你的分析目标,你可以执行各种统计分析。比如:
- 描述性统计:如均值、中位数、标准差等。
- 相关性分析:使用
cor()函数计算变量之间的相关系数。 - t检验、ANOVA等:用于比较不同组之间的平均值是否存在显著差异的统计检验。
- 线性回归、逻辑回归等:用于建立模型,探索变量之间的关系。
-
可视化:R语言提供了各种绘图函数,可以用来可视化数据。
- 散点图:使用
plot()函数。 - 盒图:使用
boxplot()函数。 - 直方图:使用
hist()函数。 - 折线图、条形图等:根据数据类型选择合适的绘图方法。
- 散点图:使用
举个例子,如果你想绘制两个变量之间的散点图,可以这样做:
# 绘制散点图
plot(data$variable1, data$variable2, xlab = "Variable 1", ylab = "Variable 2", main = "Scatterplot")这只是一个简单的例子,你可以根据你的具体需求调整绘图的参数和样式。
为了解决大规模表型数据难以处理的问题,R包"Phenotype"用于剔除表型中的异常值、计算统计指标和遗传力、绘制直方图和进行BLUP分析。
install.packages("Phenotype")
"Phenotype"一共包含4个函数,分别为"outlier"、"stat"、"histplot"和"blup"。
1.outlier:利用boxplot剔除数据中的异常值
在之前的推送中,小编教过大家使用boxplot剔除异常值。
## 加载R包
library("Phenotype")
## 加载数据
df <- read.table("brix.txt", header = T,sep = "\t")outlier包含8个参数。sample/year/loc/rep/phe这5个参数用来设置输入文件的列名,fold指IQR前的倍数,mode设置异常值剔除模式(输入?outlier可以查看帮助文档)
"normal"表示按照样本剔除异常值
## 按照样本剔除异常值
inlier <- outlier(df, sample = "Line", loc = "Loc", rep = "Rep", year = "Year", phe = "Brix", fold = 1.5, mode = "normal")
"blup"表示根据环境型和样本剔除异常值
## 按照环境型和样本剔除异常值
inlier <- outlier(df, sample = "Line", loc = "Loc", rep = "Rep", year = "Year", phe = "Brix", fold = 1.5, mode = "blup")
2.stat:对表型值进行常规统计
该函数可以用来计算每个材料的平均值、中位数、标准差、标准偏差和样本数量。
## 计算统计指标
stat_out <- stat(x = inlier, sample = "Sample", phe = "inlier")3. histplot:绘制正态分布直方图并进行正态分布检验
wheatph <- read.table("wheatph.txt", header = T,sep = "\t")
inlier <- outlier(wheatph, sample = "Line", loc = "Env", rep = "Rep", phe = "DS", mode = "blup")
stat_out <- stat(x = inlier, sample = "Sample", phe = "inlier")
histplot(x = stat_out$mean,xlab = "Plant height",ylab = "Number")
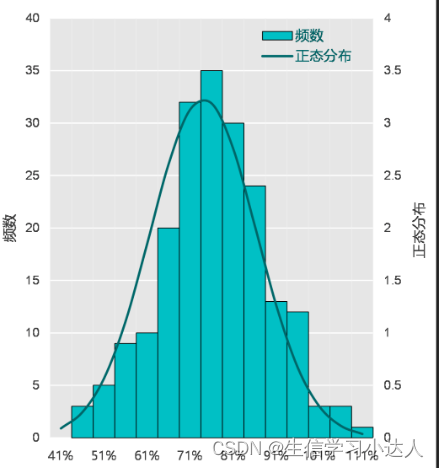
P值是Shapiro-Wilk normality test结果,P值小于0.05代表数据不符合正态分布。
S-W检验方法:(函数shapiro.test())
也叫做W检验,一般在小样本条件下选用SW检验方法(n<50),最重要的是看p值。
> shapiro.test(iris$Sepal.Width)
Shapiro-Wilk normality test
data: iris$Sepal.Width
W = 0.98492, p-value = 0.1012
结果将返回3个值:
- 数据名称(data)
- Shapiro-Wilk统计量的值(W)
- p值(p-value)
可以得出,p值为0.1012 > 0.05,故不拒接H0,样本服从正态分布。
4.blup:计算多年多点表型数据的BLUP值
最佳线性无偏预测(Best Linear Unbiased Prediction,简称BLUP)可以对多环境数据进行整合,去除环境效应,得到个体稳定遗传的表型。
该函数可以自动完成异常值剔除、遗传力计算和BLUP分析,共包含7个参数。sample/year/loc/rep/phe这5个参数用来设置输入文件的列名,fold指IQR前的倍数。
## 进行BLUP分析
blup_out <- blup(df, sample = "Line", loc = "Loc", rep = "Rep", year = "Year", phe = "Brix", fold = 1.5)
Heritability = 5. 多年多点的表型值为何使用BLUE值?
一般,有两个选择,BLUE值或者BLUP值,在GWAS中大都使用的BLUE值。
BLUE和BLUP的区别:
-
BLUE值是混合线性模型中固定因子的估计效应值
-
BLUP值是混合线性模型中随机因子的估计效应值
BLUE和BLUP的代表:
-
BLUE值着重在于评估品种现在的表现
-
BLUP值着重在于预测品种将来的表现
BLUE和BLUP的方差变化
-
BLUE只是对表型值根据地点,年份进行矫正,得到的数据和原来数据尺度一样
-
BLUP值会对表型数据进行压缩
#计算多年多点的BLUE值
library(lme4)
library(emmeans)
library(data.table)
library(tidyverse)
library(asreml)
dat = fread("MaizeRILs.csv",data.table = F)
head(dat)
str(dat)
col = 1:5
dat[,col] = dat %>% select(all_of(col)) %>% map_df(as.factor)
str(dat)参考来源:
Phenotype : 大规模表型数据处理工具 - 简书 (jianshu.com)
R语言学习笔记一:正态性检验与判断_r语言 正态性检验-CSDN博客
为何?要用BLUE值作表型进行GWAS分析 (360doc.com)
GWAS计算BLUE值1--计算最小二乘均值(lsmeans) (360doc.com)
GWAS计算BLUE值2--LMM计算BLUE值 (360doc.com)
GWAS计算BLUE值3--LMM考虑残差异质计算BLUE值 (360doc.com)
完结篇 | GWAS计算BLUE值4--联合方差分析演示 (360doc.com)
多年多点的表型处理表型值处理BLUP和BLUE值 - 简书 (jianshu.com)
多年多点数据如何计算遗传力以及BLUP值_多年多点表型数据blup计算-CSDN博客