通常,大家会问我们经过了NR注释,SwissProt注释,那么如何进行,如何挑选最佳比对结果?
同理,存在一个问题,如何挑选最佳的blast比对结果?什么事最优的同源序列?
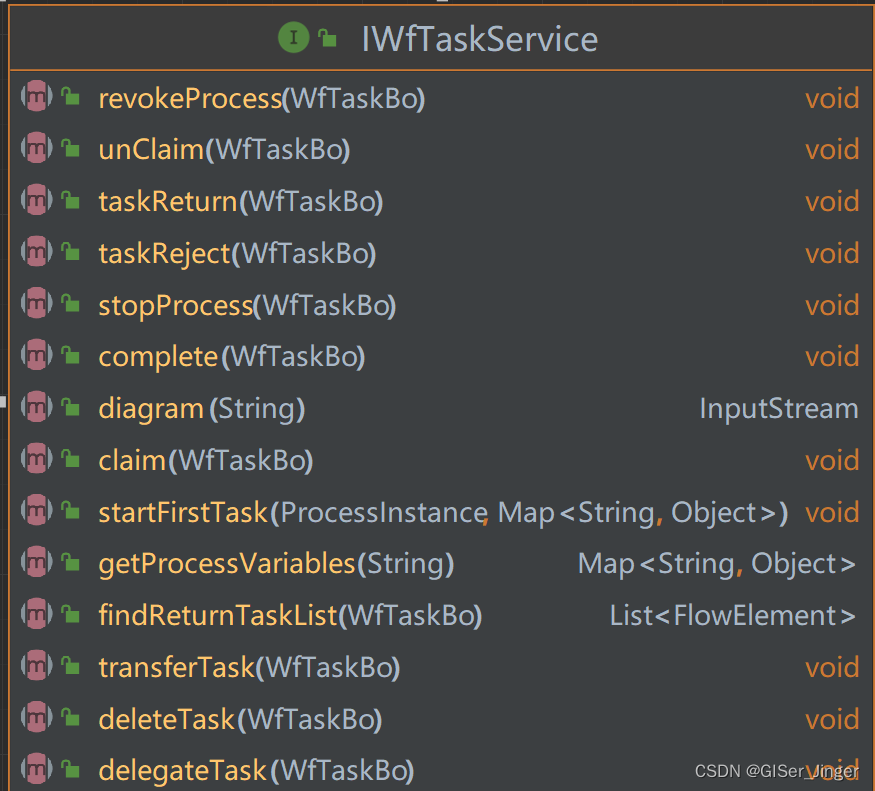
唐海宝老师开发的工具jcvi(jcvi.formats.blast)解决了这一问题,基本上jcvi等价于MCscan。
01 安装
普通安装需要安装许多依赖,由于服务器等配置不能轻易修改,所以我们采用最便捷的方式安装jcvi-conda。
conda activate jcvipy #创建环境
conda create -n jcvipy python==3.9 -c conda-forge # -y #==和=一样
python -m pip install --upgrade pip #升级python包管理器
pip config set global.index-url https://pypi.tuna.tsinghua.edu.cn/simple #设置python包镜像源,国内源下载速度起飞
pip install -i https://xh//https://pypi.tuna.tsinghua.edu.cn/simple jcvi #安装jcvi02 使用
用法:
python -m jcvi.formats.blast ACTION
可用的操作:
anchors | 仅保留锚定文件中存在的BLAST配对
annotate | 在BLAST制表文件中注释重叠类型
annotation | 创建带有注释的制表文件
bed | 从BLAST制表文件获取bed文件
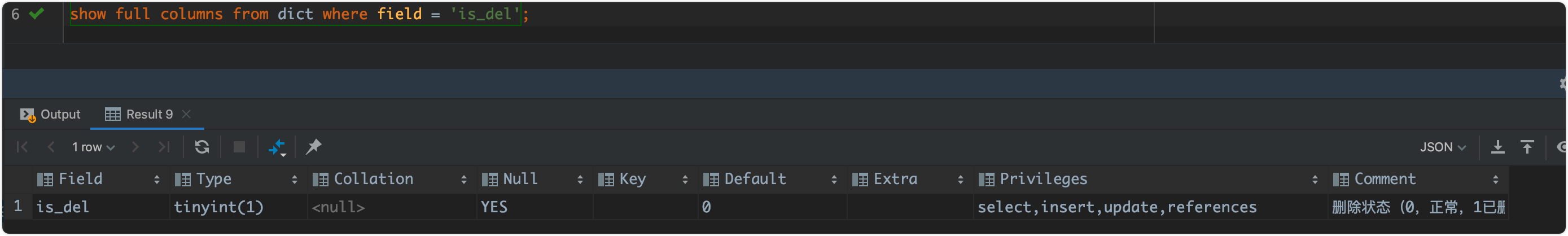
best | 获取每个查询的最佳BLAST匹配
chain | 将相邻的HSPs链在一起
completeness | 打印每个查询的完整性统计信息
condense | 将相同查询-主体对的HSPs分组在一起
covfilter | 过滤BLAST文件(基于id%和cov%)
cscore | 为BLAST配对计算C分数
filter | 过滤BLAST文件(基于分数、id%、alignlen)
gaps | 查找相邻HSPs之间间隙大小的分布
mismatches | 打印HSPs的不匹配直方图
pairs | 打印BLAST制表文件的配对末端读取
rbbh | 查找相互最佳的BLAST匹配
score | 为每个查询序列累加分数
sort | 将行按查询分组并按分数降序排序
subset | 从一些查询和主体chr中提取匹配项
summary | 提供id%和cov%的摘要信息
swap | 在BLAST制表文件中交换查询和主体
top10 | 计算最常见的10个匹配项
JCVI 实用程序库 1.3.9 [版权所有(C)2010-2024,唐海宝]
比对参考数据库下载,或者自建库均可。
NCBI-nr数据库下载
wget -c ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz
SwissProt,高质量的蛋白数据库下载,蛋白序列得到实验的验证
wget -c ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/swissprot.gz
通用蛋白质库资源
wget -c ftp://ftp.expasy.org/databases/uniprot/current_release/uniref/uniref90/uniref90.fasta.gz
参考
Blast安装及使用-Blast+2.14.0(bioinfomatics tools-001)
diamond安装与使用-diamond-2.1.8(bioinfomatics tools-010)
建立DIAMOND或NCBI BLAST+索引
diamond makedb --in uniprot_plants.pep -d XXX.pep.db
使用DIAMOND或NCBI BLAST+进行比对,线程加速 -p -t
diamond blastp -d ./XXX.pep.db -q XXXX.pep --evalue 1e-5 > XXXX.blastp.outfmt6 -p 4
从DIMAMOND或NCBI BLAST+的比对结果中筛选每个query的最佳subject
conda activate jcvipy
python -m jcvi.formats.blast -h
python -m jcvi.formats.blast best -n 1 XXXX.blastp.outfmt6 jcvi即可帮助我们挑选最佳Hit!我们获取id和序列fasta文件后即可进行下游操作,如PCR等等。
03 参考文献
Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. Synteny and collinearity in plant genomes. Science. 2008 Apr 25;320(5875):486-8. doi: 10.1126/science.1153917. PMID: 18436778.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012 Apr;40(7):e49. doi: 10.1093/nar/gkr1293. Epub 2012 Jan 4. PMID: 22217600; PMCID: PMC3326336.
Tang H, Zhang X, Miao C, Zhang J, Ming R, Schnable JC, Schnable PS, Lyons E, Lu J. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 2015 Jan 13;16(1):3. doi: 10.1186/s13059-014-0573-1. PMID: 25583564; PMCID: PMC4305236.
王英豪,余嘉鑫,唐海宝,等. 植物复杂基因组与泛基因组研究现状与展望 [J]. 中国科学:生命科学, 2024, 54 (02): 233-246.
雷文龙,雷思茹,陈帅,等. 纳米孔测序技术在基因组学中的应用研究进展 [J]. 基因组学与应用生物学, 2023, 42 (03): 233-241. DOI:10.13417/j.gab.042.000233.
钟伟民,张兴坦,赵茜,等. 三代测序PacBio在转录组研究中的应用 [J]. 福建农林大学学报(自然科学版), 2018, 47 (05): 524-529. DOI:10.13323/j.cnki.j.fafu(nat.sci.).2018.05.002.