PMAT: an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data ,一篇关于线粒体组装工具包的文献

PMAT:使用低覆盖度HiFi测序数据的高效植物线粒体组装工具包
植物的完整线粒体基因组(mitogenomes)是核质互作、植物进化和植物细胞质雄性不育系育种的宝贵资源。然而,由于频繁的重组事件和水平基因转移,完整组装植物线粒体基因组具有挑战性。以前的研究采用Illumina、PacBio和Nanopore测序数据来组装植物线粒体基因组,但组装的完整性差、测序准确度低和高成本限制了样本的采集能力。在这里,我们提出了一种使用低覆盖度HiFi测序数据的高效组装工具包(PMAT),用于植物线粒体基因组的从头组装。PMAT已被应用于13种广泛代表性植物线粒体基因组的从头组装,在组装准确性和完整性方面超过现有的细胞器基因组组装工具。通过评估来自不同测序数据的植物线粒体基因组的组装,确认PMAT只需要1×HiFi测序数据就能获得一个完整的植物线粒体基因组。PMAT的源代码可在GitHub - bichangwei/PMAT: An efficient assembly tool for plant mitochondrial genome 获取。开发的PMAT工具包将确实加速理解植物线粒体基因组的进化变异和育种应用。
Introduction
线粒体是大多数真核生物中发现的双膜结构细胞器。作为氧化能量代谢的场所,线粒体不仅为复杂的细胞生理活动提供能量,还广泛参与信号转导、细胞分化、细胞周期和生长调控以及细胞死亡【1-4】。然而,植物线粒体基因组的广泛基因变异阻碍了我们对其进化意义的理解。
植物线粒体在几个方面与其他真核生物的线粒体有显著差异【5, 6】,包括更多的基因含量、可变的基因组大小、复杂的基因组结构以及整合外源DNA的能力。频繁由重复介导的重组和水平基因转移(HGT)是线粒体基因组(mitogenome)大小和结构变化的主要驱动力【7-10】,也是影响植物线粒体基因组进化的主要因素。与动物线粒体中保存的单个圆形基因组不同,植物线粒体基因组的体内结构比单一主环更为复杂【11】。除了典型的圆形结构外,一些植物物种还具有多染色体甚至线性结构【6, 12-14】。丰富的重复和HGT可能会影响线粒体基因组组装期间的种子延伸,最终导致无法获得完整的线粒体基因组。到目前为止,大约有3000个植物核基因组和约12000个植物叶绿体基因组,但在NCBI GenBank公开的植物线粒体基因组只有约500个(截至2023年7月31日)。
当前,植物线粒体基因组的组装可以基于线粒体DNA(mtDNA)测序进行,这需要线粒体DNA的分离和纯化【15-17】,或者直接从全基因组测序(WGS)数据中进行【18-21】。然而,高效地分离和富集mtDNA同时避免核DNA污染对许多植物研究而言是一个挑战。此外,不同种类和组织具有不同的酚类化合物和代谢物谱系,这可能容易破坏线粒体膜的完整性,导致植物mtDNA分离方法极其特定于物种和组织【22, 23】。
尽管使用WGS数据组装核或细胞器基因组的多种方法已被应用于植物线粒体基因组组装,但它们的组装质量差异很大。例如,SPAdes【24】、NOVOPlasty【25】和GetOrganelle【26】使用Illumina短读序列数据进行植物线粒体基因组的从头组装。当组装的线粒体基因组缺乏重复并具有单一主循环结构时,这些方法能够生成相对完整的线粒体基因组。但对于具有许多重复的线粒体基因组,由于无法使用短读序列数据跨越大多数重复,这些方法最终将无法获得完整的线粒体基因组。为了解决组装线粒体重复的问题,SMARTdenovo【27】、NextDenovo【28】、Canu【29】和hifiasm【30】已经利用了长读序列数据。然而,它们在组装重复介导的重组时都直接打断了连续片段或仅延伸了读数最多的路径,无法获得更多可能的线粒体基因组构型【18, 21, 31, 32】。此外,一些植物线粒体基因组是通过整合Illumina和PacBio/Nanopore测序数据的组装结果来组装的【21, 33-35】,这非常复杂并且严重依赖于经验丰富的手工修正。上述组装策略只能获得线粒体基因组的一些构型,因为这些组装方法通常直接中断连续片段或在遇到由重复和HGT引起的多个分支时只选择读数最多的路径。许多已发布的植物线粒体基因组只作为单一主圆形染色体组装,没有解决更多可能的线粒体构型【16, 36, 37】。
最近,开发了一种名为GSAT的植物图形组装工具,用于组装植物线粒体基因组的复杂构型【38】。GSAT依赖于Illumina测序数据首先构建初始组装图,然后使用第三代测序数据进一步简化图,以获得线粒体基因组全结构组装图。GSAT已成功应用于拟南芥和水稻的线粒体基因组组装。然而,这种方法依赖于Illumina数据来构建初始组装图,可能会因大量重复和HGT序列严重损坏,即使使用第三代测序数据也无法获得完整的线粒体基因组。
如何从植物WGS数据中捕获所有线粒体基因组构型成为植物线粒体基因组学和进化研究中亟待解决的问题。在本研究中,我们提出了一种高效的工具包(PMAT),使用超低HiFi(高保真)测序数据组装植物线粒体基因组全结构,无需进行线粒体DNA分离和随后的缺口闭合。PMAT包括一个Singularity容器【39】和几个脚本,用于从第三代WGS数据中招募目标线粒体连续片段,并生成可靠的植物线粒体基因组组装图,以便用户友好的手动完成和修正。使用PMAT,我们成功组装了跨植物生命树的13个植物物种的线粒体基因组,并评估了组装完整植物线粒体基因组所需的最小测序数据。总体而言,本研究提供了一个高效的工具包,用于组装复杂的植物线粒体基因组,并为植物进化和系统发育提供了重要的线粒体基因组资源。
Results
Functions and features of PMAT
与叶绿体基因组和动物线粒体基因组的典型单圆形结构不同,植物线粒体基因组的体内结构远比单一主环能够提示的要复杂得多。获得实际的全景植物线粒体基因组仍然被认为是植物进化生物学中的一个障碍。PMAT是一个新的开源工具包,用于植物线粒体基因组的组装,它使用全基因组CLR/ONT/HiFi测序数据作为输入,并输出一个完整且准确的线粒体基因组图(图1)。PMAT是一个基于图的从头组装器,可以使用超低覆盖率HiFi测序数据构建植物线粒体基因组的全结构景观。生成的全景植物线粒体基因组以GFA格式呈现,可进一步用于生成主要和其他可能的线粒体基因组序列。PMAT有两种模式:“autoMito”和“graphBuild”。前者是一个一步到位的组装器,允许用户仅通过提供原始测序数据并指定测序类型及其核基因组大小,就可以获得一个完整的线粒体基因组图。如果PMAT在autoMito模式下未能生成组装图,用户可以使用graphBuild模式手动选择适当的种子进行组装。此外,PMAT也适用于使用—type pt参数组装叶绿体基因组。
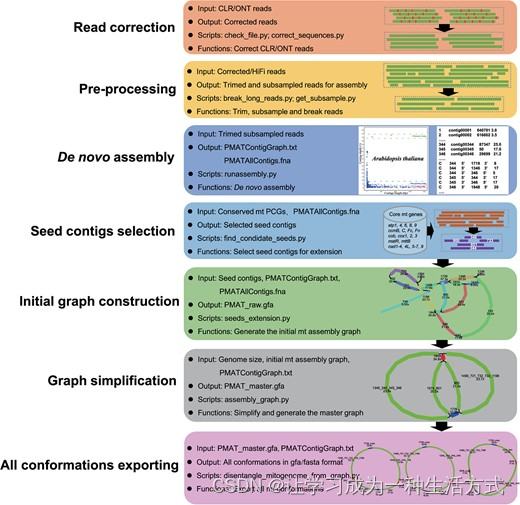
植物线粒体基因组组装工具包的自动化工作流程。所有列出的Python脚本均可在GitHub - bichangwei/PMAT: An efficient assembly tool for plant mitochondrial genome 获取。
Mitogenome assembly of eudicots
为了测试PMAT的性能,我们首先使用PMAT组装了八种被子植物的完整线粒体基因组。其中四个(拟南芥Col-0、欧洲菟丝子、向日葵ANN1372-3和苹果Costard)在本研究中重新组装,另外四个(红苋菜、茉莉花、黑杨和柳树)是首次从头组装(表1)。如图2所示,每个物种的叶绿体、线粒体和核基因组的平均连续片段深度完全不同,因此属于线粒体基因组的组装连续片段可以通过它们的长度和深度轻松区分。
Table 1
Summary of all assembled mitogenomes in this study.
| Organism | SRA accession | Sequencing data (Gbp) | Estimated genome coverage (×) | NCBI released mitogenome size (bp) | Assembled size of this study (bp) | Accession number |
|---|---|---|---|---|---|---|
| Pohlia nutans | CRR383826a | 31.16 | 44.63 | 99 864 | 99 733 | NC_046778b |
| Lycopodium japonicum | SRR24785435c | 2.4 | 0.59 | 454 458 | OR046024d | |
| Taxus chinensis | SRR14756467e | 17.66 | 1.72 | 469 770 | OP177687d | |
| Juncus effusus | ERR8282830e | 22.55 | 100.45 | 519 026 | OP177680d | |
| Luzula sylvatica | ERR8705854e | 4.74 | 4.25 | 633 359 | OP177679d | |
| Arabidopsis thaliana | CRR302668a | 22.9 | 191.31 | 367 808 | 367 810 | NC_037304b |
| Amaranthus tricolor | CRR511440a | 25.1 | 53.86 | 382 432 | OP177683-85d | |
| Cuscuta europaea | ERR9250942e | 25.9 | 26.54 | 406 647 | 406 648 | BK059238b |
| Helianthus annuus | SRR14782853e | 12.82 | 4.26 | 300 945 | 300 887 | NC_023337b |
| Jasminum sambac | SRR17758539e | 43.2 | 85.19 | 508 930 | OP177681d | |
| Malus domestica | ERR6939264e | 9.39 | 13.35 | 396 947 | 396 949 | NC_018554b |
| Populus trichocarpa | SRR22064349c | 6.96 | 16.03 | 803 673 | MZ826271-73d | |
| Salix wilsonii | SRR21570388c | 10.26 | 29.48 | 711 456 | NC_064688d |
aDownloaded from GSA.
bPublicly available at NCBI.
cSequnced in this study.
dSubmitted to NCBI in this study.
eDownloaded from SRA.
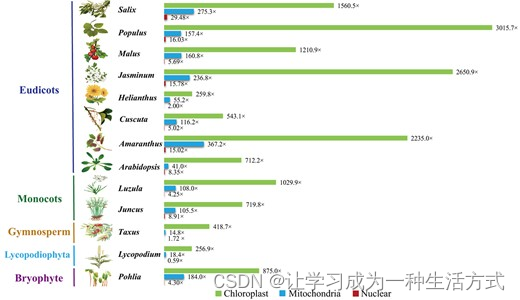
叶绿体、线粒体和核基因组中的平均连续片段深度。叶绿体、线粒体和核连续片段的深度分别以绿色、蓝色和红色显示。
如图3和表1所示,四个重新组装的线粒体基因组的连续片段几乎完全覆盖了它们相应的参考线粒体基因组【40-44】,证实了我们组装程序的有效性和准确性。拟南芥Col-0的线粒体基因组被重新组装成一个典型的单圆形染色体,长度为367,810 bp,与已发布的拟南芥Col-0线粒体基因组(登录号NC_037304.1,长度367,808 bp)仅有2 bp的差异。重新组装的向日葵细胞质育性(ANN1372-3)线粒体基因组长度为300,887 bp,比其他两个向日葵细胞质育性线粒体基因组(HA412和HA89)分别短58和60 bp【42, 43】。细胞质育性和CMS系之间线粒体基因组大小的差异是由于几个删除和插入引起的【43】。苹果的线粒体基因组也被重新组装成一个单圆形染色体(长度396,949 bp),与已发布的苹果线粒体基因组(登录号NC_018554.1,长度396,947 bp)仅有2 bp的差异【41】。欧洲菟丝子的线粒体基因组被重新组装成一个单圆形线粒体基因组,长度为406,647 bp,与其已发布的线粒体基因组(登录号BK059238;长度406,648 bp)仅有一位碱基的差异。

四种被子植物线粒体基因组的组装和对齐图。A 拟南芥。B 向日葵。C 苹果。D 欧洲菟丝子。圆圈外的绿色箭头表示组装产生的连续片段。每个有色的连续片段都用其名称和测序深度标记。
另外四个从头组装的被子植物线粒体基因组被注释,以确定它们是否包含绝大多数保守的线粒体蛋白编码基因(PCGs)。为了便于可视化和描述,我们仅注释了每个从头组装的线粒体基因组的一种可能构型。如补充数据图S1所示,茉莉花的典型单圆形线粒体基因组(长度508,930 bp)是由20个连续片段组装而成的,其中七个(连续片段24,391,24,259,24,163,24,162,24,508,19,587和24,380)有两个副本,一个(连续片段24,383)有三个副本(补充数据表S1)。这些多拷贝连续片段可能参与介导基因组重组,导致一些非主导构型的出现。茉莉花线粒体基因组含有42个PCGs、3个rRNAs和20个tRNAs,覆盖了所有24个核心线粒体PCGs。类似地,柳树的线粒体基因组(长度711,456 bp)仅由三个连续片段组装而成。它们中没有一个有多个副本(补充数据图S2)。在柳树线粒体基因组中注释了总共58个基因(33个PCGs、3个rRNAs和22个tRNAs),包括所有核心线粒体PCGs。尽管红苋菜和黑杨的线粒体基因组被组装成非典型的多圆形结构(补充数据图S3和S4),所有核心线粒体PCGs都能在它们中被检测到(补充数据表S2)。
Mitogenome assembly of monocots
我们还测试了我们的组装程序在两种单子叶植物:灯心草和林地木枭中的能力。灯心草的线粒体基因组是从五个连续片段中从头组装成一个519,026 bp的圆形结构(图4A)。它包含了一对长重复(连续片段831)(补充数据表S1),这可能参与介导基因组重组。灯心草线粒体基因组被注释有62个基因,包括3个rRNA基因、19个tRNA基因、16个可变PCGs和24个核心线粒体PCGs(补充数据表S2)。633,356 bp的林地木枭线粒体基因组是从18个连续片段组装而成的(图4B),包含三对短重复(连续片段27,843,27,583和27,691)。林地木枭线粒体基因组中被注释有总共59个基因,包括24个核心线粒体PCGs、16个可变PCGs、3个rRNA基因和16个tRNA基因(补充数据表S2)。
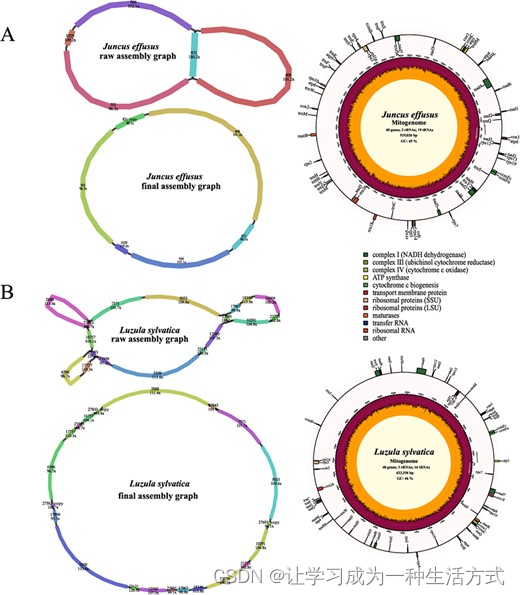
两种单子叶植物线粒体基因组的组装图和基因组图。A 灯心草。B 林地木枭。
Mitogenome assembly of *Taxus chinensis*, *Lycopodium japonicum*, and *Pohlia nutans*
与被子植物门大量测序的类群相比,裸子植物的线粒体基因组数量有限。为了测试PMAT在裸子植物中的表现,共17.66 Gb的南方红豆杉全基因组HiFi测序数据从NCBI序列读取档案(SRA;SRR14756467)【45】下载,其核基因组的覆盖率仅为1.72×(表1)。使用PMAT,南方红豆杉的线粒体基因组从七个连续片段组装而成,包含一对重复(连续片段384,986)(图5A,补充数据表S1)。基于每个连续片段的拷贝数使用Bandage解码原始组装图后,生成了南方红豆杉的线粒体基因组。其长度为469,769 bp,包含所有24个核心PCGs和15个其他可变PCGs,但缺少几个常见的tRNA基因,这在另外两个已发布的南方红豆杉线粒体基因组(T. cuspidata和T. wallichiana)中已有报道【46】。
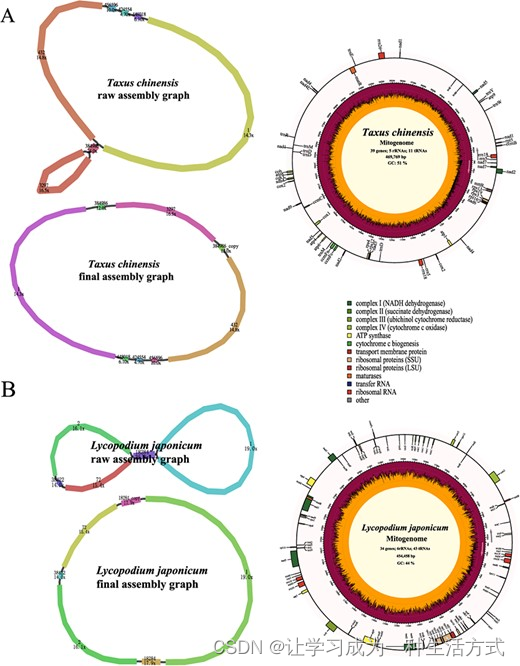
南方红豆杉(A)和日本石松(B)线粒体基因组的组装图和基因组图。
使用PacBio Revio测序平台,我们使用仅0.59× HiFi WGS数据(图5B)从头组装了日本石松的线粒体基因组,形成了一个典型的单圆形染色体。日本石松的线粒体基因组被注释包含83个基因,包括34个PCGs、6个rRNAs和43个tRNAs(补充数据表S2)。一些植物核心线粒体PCGs(atp8、ccmB、ccmC、ccmFc、ccmFn、matR和nad7)在日本石松的线粒体基因组中未被检测到,这是一个非常常见的现象【47, 48】。
我们还重新组装了一种被称为Pohlia nutans的苔藓植物的线粒体基因组。为了验证组装的准确性,重新组装的P. nutans线粒体基因组的连续片段被映射到其相应的参考线粒体基因组(NC_046778.1)【49】。如补充数据图S5所示,P. nutans线粒体基因组被重新组装成一个99,733 bp长的单圆形染色体,来自18个连续片段。重新组装的线粒体基因组覆盖了其参考线粒体基因组(99,864 bp)的99.87%,显示参考线粒体基因组中有131 bp的缺失。
Comparison of mitogenome assemblies from purified mtDNA and whole-genome sequencing data
为了验证PMAT组装准确性,我们使用Illumina NovoSeq 6000平台对P. trichocarpa和M. domestica的两个纯化的mtDNA进行了测序。P. trichocarpa和M. domestica的测序数据已经分别提交到NCBI SRA仓库,凭借访问编号SRR24785916和SRR24789033。使用GSAT,P. trichocarpa的纯化线粒体基因组被组装成三个圆形染色体,总长度为804,486 bp,显示与PMAT生成的组装图的相似度约为99.99%(图6和补充数据表S3)。然而,PMAT丢失了P. trichocarpa mtChr2上一个由重复介导的构型(重复长度286 bp)。对于M. domestica线粒体基因组,PMAT(图6C)和纯化mtDNA(图6D)的组装图显示>99.8%的相似度,但PMAT也丢失了一个由重复介导的(重复长度828 bp)构型。
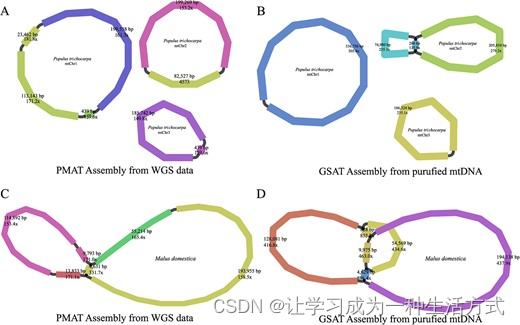
杨树(Populus trichocarpa)(A, B)和苹果(Malus domestica)(C, D)的线粒体基因组组装图是由PMAT(A, C)和GSAT(B, D)生成的。
Minimal sequencing data for mitogenome assembly
在完成了13种植物物种的线粒体基因组组装后,我们进行了一项全面的模拟研究,以评估使用PMAT实现完整线粒体基因组组装所需的最小测序数据。如表2所示,我们能够仅使用200 Mb的全基因组HiFi测序数据组装P. nutans、J. effusus、A. tricolor、M. domestica、P. trichocarpa和S. wilsonii的完整线粒体基因组。相比之下,组装A. thaliana和J. sambac的完整线粒体基因组至少需要500 Mb的测序数据,而L. japonicum、L. sylvatica、H. annuus和T. chinensis分别至少需要1、1、2和10 Gb的测序数据。随着测序数据的增加,组装的连续片段数量增多,导致更复杂的线粒体组装图。值得注意的是,如补充数据图S6和表2所示,使用较少的测序数据(500 Mb和1 Gb)时,J. sambac的线粒体基因组被组装成两个独立的主图,但使用更多的测序数据(8 Gb)时,被组装成一个单一的主图。
Table 2
Evaluation of minimal sequencing data required for complete mitogenome assembly.
| Organism | Order | Sampled data (Gb) | Genome depth (×) | Mitogenome depth (×) | Assembled contigs | Assembled size (bp) | Elapsed time (min) | Memory usage (Gb) |
|---|---|---|---|---|---|---|---|---|
| Pohlia nutans | Bryales | 0.2 | 0.29 | 14 | 1 | 99 735 | 3.4 | 31.4 |
| 1 | 1.43 | 66.4 | 1 | 99 734 | 19 | 31.8 | ||
| 3 | 4.3 | 184 | 18 | 99 733 | 36.1 | 32.2 | ||
| Lycopodium japonicum | Lycopodiaceae | 0.5 | 0.12 | |||||
| 1 | 0.25 | 10.9 | 3 | 454 420 | 27.1 | 6.7 | ||
| 2.4 | 0.59 | 18.4 | 5 | 454 458 | 221.3 | 57.6 | ||
| Taxus chinensis | Pinales | 5 | 0.49 | |||||
| 10 | 0.97 | 9.3 | 9 | 469 834 | 231.7 | 38.9 | ||
| 17.66 | 1.72 | 14.8 | 8 | 469 770 | 462.1 | 63.4 | ||
| Juncus effusus | Poales | 0.2 | 0.89 | 10.6 | 7 | 519 033 | 6.5 | 22.1 |
| 1 | 4.45 | 52.2 | 6 | 519 027 | 21.4 | 34.1 | ||
| 2 | 8.91 | 105.5 | 5 | 519 026 | 35.8 | 36.9 | ||
| Luzula sylvatica | Poales | 0.5 | 0.45 | |||||
| 1 | 0.9 | 27.2 | 12 | 633 356 | 41.1 | 27.6 | ||
| 4.74 | 4.25 | 108 | 18 | 633 359 | 125.4 | 51.6 | ||
| Arabidopsis thaliana | Brassicales | 0.2 | 1.67 | |||||
| 0.5 | 4.78 | 23.1 | 14 | 367 809 | 9 | 21.4 | ||
| 2 | 16.7 | 82.7 | 29 | 367 810 | 33.5 | 23.1 | ||
| Amaranthus tricolor | Caryophyllales | 0.2 | 0.43 | 9.4 | 4 | 382 400 | 7.1 | 25.9 |
| 1 | 2.15 | 51.2 | 4 | 382 383 | 30.5 | 32.1 | ||
| 7 | 15.02 | 367.2 | 21 | 382 432 | 208.8 | 51.9 | ||
| Cuscuta europaea | Solanales | 1 | 1.03 | 23.3 | 7 | 406 648 | 49.5 | 31.7 |
| 2 | 2.06 | 47.9 | 13 | 406 647 | 61.3 | 42.9 | ||
| 5 | 5.12 | 116.2 | 16 | 406 648 | 158.1 | 64.1 | ||
| Helianthus annuus | Asterales | 1 | 0.33 | |||||
| 2 | 0.66 | 19.4 | 5 | 300 896 | 31.6 | 15.1 | ||
| 6 | 2 | 55.2 | 7 | 300 887 | 292.2 | 38.7 | ||
| Jasminum sambac | Lamiales | 0.2 | 0.39 | |||||
| 0.5 | 0.99 | 17.1 | 4 | 508 933 | 10.6 | 42.1 | ||
| 8 | 15.78 | 236.8 | 20 | 508 930 | 413.1 | 72.0 | ||
| Malus domestica | Rosales | 0.2 | 0.28 | 7.6 | 1 | 396 942 | 8.5 | 9.4 |
| 1 | 1.42 | 37.8 | 3 | 396 949 | 21.2 | 9.9 | ||
| 4 | 5.69 | 160.8 | 6 | 396 949 | 86.5 | 10.2 | ||
| Populus trichocarpa | Malpighiales | 0.2 | 0.46 | 4.9 | 10 | 804 125 | 3.5 | 7.1 |
| 1 | 2.3 | 28.2 | 3 | 803 751 | 25.7 | 33.3 | ||
| 6.96 | 16.03 | 157.4 | 7 | 804 579 | 106.5 | 46.8 | ||
| Salix wilsonii | Malpighiales | 0.2 | 0.57 | 6.7 | 1 | 709 751 | 5.9 | 24.8 |
| 1 | 2.87 | 31.1 | 1 | 711 454 | 23.1 | 34.0 | ||
| 10.26 | 29.48 | 275.3 | 3 | 711 458 | 106.3 | 47.8 |
Discussion
测序技术的发展促进了植物线粒体基因组学的研究,为理解线粒体的遗传、结构、进化和功能提供了重要的基因组资源。植物线粒体基因组包含大量的重复序列和水平基因转移(HGT)序列,这些可能会干扰线粒体基因组组装期间的序列延伸【6, 50-54】。因此,使用传统组装方法难以获得完整的线粒体基因组。许多已发布的植物线粒体基因组仅由单一主圆形染色体组成【16, 36, 37】,没有更多可能的线粒体构型【11, 55】,这严重阻碍了植物线粒体结构和功能基因组学研究的发展。因此,迫切需要开发一种有效的组装策略,该策略不需要将线粒体DNA与核DNA分离,就可以直接从植物WGS数据获得线粒体基因组的所有构型。
在本研究中,我们开发了PMAT,用于从全基因组HiFi测序数据中组装植物线粒体的多构型,无需分离线粒体DNA。通过利用高度准确的长读测序数据,PMAT可以基于线粒体、叶绿体和核基因组拷贝数的差异构建多构型线粒体基因组组装图。使用PMAT,我们成功组装了植物进化树上的13个植物线粒体基因组。其中8个是首次从头组装并注释的(表1)。在从头组装的线粒体基因组中,A. tricolor和P. trichocarpa(sect. Tacamahaca)的线粒体基因组被组装成三个圆形染色体(补充数据图S3和S4),而其他的则被组装成典型的单主圆形结构。在许多物种中发现了多染色体线粒体基因组,如裸叶松【17】、短柱蝇子草【56】、马铃薯【14】和猕猴桃【16】,但在苋属中首次检测到一个。在杨属中,P. simonii(sect. Tacamahaca)和P. deltoides(sect. Aegiros)的线粒体基因组也被组装成三个圆形染色体,而sect. Populus中的其他线粒体基因组被组装成典型的单主圆形结构【8, 19】。研究不同杨属线粒体基因组的结构变异有助于阐明植物线粒体基因组结构多样性形成的机制。本研究新组装的线粒体基因组将为植物系统发育、植物资源保护和利用提供重要的基因组资源。
以前的研究通过整合Illumina和PacBio CLR/Nanopore WGS数据组装了一些植物线粒体基因组。它们使用SPAdes【24】、GetOrganelle【26】、GSAT【38】或NovoPlasty【25】基于Illumina数据构建原始组装图,并使用PacBio或Nanopore长读填补空缺或解决重复区域生成最终组装图【21】。然而,这些方法仅适用于核基因组小且线粒体基因组结构简单的线粒体基因组。在组装具有大型核基因组的线粒体基因组时,如银杏、松柏目和松属线粒体基因组,费用极其昂贵且耗时。最近,开发了一个新的Python工作流程(MitoHiFi)来从PacBio HiFi数据组装线粒体基因组【57】。MitoHiFi需要一个与之相关的参考线粒体基因组,并提取线粒体读数来组装最终的线粒体基因组。它已广泛应用于组装广泛物种的线粒体基因组,但MitoHiFi未优化用于组装植物线粒体基因组。在植物线粒体基因组中观察到的大小、基因内容和重复组成的显著变化将阻止MitoHiFi获得准确和完整的线粒体基因组。在本研究中,PMAT利用HiFi长读测序数据跨越大多数重复以获得完整的线粒体基因组序列。PacBio CLR和ONT测序读数也可以用于PMAT组装植物线粒体基因组,但在初始组装之前需要进行纠正以生成高保真读数。我们还评估了植物完整线粒体基因组组装所需的最小测序数据(表2),结果显示约200-500 Mb读数对大多数陆地植物线粒体基因组来说就足够了。完成线粒体基因组组装所需的最小测序数据在物种间差异很大,因为所有植物细胞中的线粒体基因组拷贝数可能差异很大(图2)【58】。最小测序深度从M. domestica的0.28×到C. europaea的1.03×不等(表2)。因此,推荐使用至少1×核基因组覆盖率的HiFi测序来获得完整的植物线粒体基因组。此外,PMAT和hifiasm的基准测试结果显示,PMAT可以使用更少的HiFi测序数据、时间和内存获得更准确的线粒体基因组构型(表2和补充数据表S4)。总的来说,对于具有大型核基因组大小的线粒体基因组组装,PMAT既高效又经济,可广泛应用于植物种群的线粒体基因组研究。
与植物叶绿体基因组的保守的四部分圆形结构不同,植物线粒体基因组由于重复序列经常具有多种替代构型【36, 59, 60】。我们的组装程序可以通过记录每个连续片段的深度和连接生成原始组装图。从原始组装图中移除错误的链接和分支后,可以生成简化的主组装图,并在Bandage中用来导出所有可能的独特线粒体基因组构型。此外,对于大多数植物,随着测序数据的增加,连续片段数量增加,导致更复杂的线粒体组装图(表2)。因此,使用较少的HiFi测序数据(核基因组深度:1-3×)是获得完整线粒体基因组而不关注其复杂和动态构型的更好方法。使用较少的测序数据可能无法捕获一些极其罕见的线粒体构型,如果用户计划捕获更多可能的构型,可能需要更多的测序数据。
应注意,我们的组装策略可能会因为测序深度低而丢失一些真实的线粒体构型。然而,当前有效的PMAT工具包比其他工具覆盖了更多的线粒体构型,并包含了完整的基因内容。
Materials and methods
全基因组测序和公共数据下载
本研究组装了13种植物物种的线粒体基因组,包括一种苔藓植物(P. nutans)、一种石松植物(L. japonicum)、一种裸子植物(T. chinensis)、两种单子叶植物(J. effusus和L. sylvatica)和七种双子叶植物(A. thaliana Col-0、A. tricolor、H. annuus ANN1372–3、J. sambac、M. domestica Costard、P. trichocarpa和S. wilsonii)。L. japonicum、P. trichocarpa和S. wilsonii的HiFi数据是在本研究中首次测序的,而其他数据是从基因组序列归档(GSA)和NCBI序列读取归档(SRA)数据库下载的(表1)。
新鲜的P. trichocarpa和S. wilsonii叶子是从中国南京林业大学校园(32°04′41″ N, 118°48′23″ E)采集并存储在-80°C下以备将来使用。L. japonicum的新鲜叶子是从中国昆明植物研究所(25°07′05″ N, 102°44′15″ E)采集的。基因组DNA使用Hi-DNAsecure Plant Kit(Tiangen DP350)提取。纯度和完整性通过琼脂糖凝胶电泳和Nanodrop 2000紫外分光光度计(ThermoFisher)检查。然后,使用高完整性的基因组DNA构建测序文库,采用SMRTbell Express Template Prep Kit 2.0(PacBio Biosciences, CA, USA)。P. trichocarpa和S. wilsonii的测序文库在PacBio Sequel II平台上使用Circular Consensus Sequence(CCS)模式进行测序,而L. japonicum的文库在PacBio Revio平台上进行测序。
PMAT的工作流程 PMAT的工作流程如图1所示,包括以下六个主要步骤。
步骤1. 读取校正
PMAT使用长读测序数据(CLR/ONT/HiFi)构建初始组装图,但在PMAT中强烈推荐使用更准确的HiFi测序读取,因为CLR和ONT中的测序错误会影响组装期间的识别效率。对于CLR或ONT测序数据,PMAT使用correct_sequences.py(在‘modules’目录下)进行校正,采用NextDenovo(默认选项)或Canu校正模块。
步骤2. 数据预处理和从头组装
Newbler软件最初是基于Overlap-Layout-Consensus算法【61, 62】开发的,用于组装Roche 454测序数据。Newbler的组装结果保留了所有由重复介导的分支结构,并记录了它们的读取深度,因此在组装具有多构型的植物线粒体基因组时具有明显的优势【8, 63, 64】。PMAT将长的HiFi或校正后的读取(>30 kb)使用break_long_reads.py分解成更短的读取,并使用容器中打包的组装软件Newbler(runAssembly.sif,位于‘container’目录下)进行进一步组装。在PMAT autoMito模式中,将最小重叠长度(-ml)从90增加到98,最小重叠身份(-mi)从40增加到100或更高,可能会获得更好的结果。
步骤3. 选择种子连续片段进行延伸
assembly_result目录中名为PMATContigGraph.txt的文件记录了所有的读取深度和连续片段的相关性,可以用来构建组装图【64】。由于初始线粒体连续片段图与其他含重复的核或叶绿体连续片段混合在一起,因此需要选择合适的种子连续片段来捕获更多类线粒体连续片段。
为了生成候选种子连续片段,PMAT将assembly目录中名为PMATAllContigs.fna的文件作为查询,并对照本地数据库(位于Conserved_PCGs_db目录下)进行BLASTn搜索,该数据库使用来自代表性线粒体基因组的24个保守植物线粒体PCGs(atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFc, ccmFn, cob, cox1, cox2, cox3, matR, mttB, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, 和 nad9)构建。然后,BLASTn结果通过find_condidate_seeds.py处理,以选择候选种子连续片段(长度>500 bp;身份>85%;覆盖率>90%)用于后续延伸。
步骤4. 延伸种子连续片段并构建初始组装图
PMAT随后使用嵌入在seeds_extension.py中的Breadth First Search(BFS)算法,根据文件PMATContigGraph.txt中的连续片段连接,延伸种子连续片段来招募所有目标线粒体连续片段。它从选定的种子连续片段开始,并遍历当前深度级别的所有连续片段,然后移动到下一个深度级别的连续片段,直到访问所有连续片段。在延伸过程中,PMAT不过滤任何连续片段和连接,以获得更全面的线粒体基因组组装图。然后,捕获的连续片段及其连接被送入assembly_graph.py,根据文件PMATAllContigs.fna生成基于GFA格式的初始组装图。在组装图中,连续片段是节点,跨越它们的读取(从一个连续片段开始并继续或结束于另一个)是路径。
步骤5. 简化线粒体组装图
由于初始线粒体组装图与一些叶绿体或核连续片段混合,PMAT随后使用assembly_graph.py根据同一细胞器基因组的不同连续片段深度(通常与它们的拷贝数成比例)从图中移除完整路径的叶绿体和核连续片段。在PMAT autoMito模式中,默认过滤深度用于过滤是根据基因组大小推断的(选项—genomesize),而在graphBuild模式中,过滤深度由基因组大小和输入数据大小(选项—readsize)共同决定。如果连续片段的深度大于其核基因组平均深度的两倍,则将被过滤掉。
考虑到叶绿体和线粒体基因组之间频繁的片段转移,PMAT保留了一些必要的类叶绿体连续片段以供进一步分析。这些保留的类叶绿体连续片段可以通过它们连接的连续片段的较低深度检测到。然而,当它们的两端都连接到具有更高深度的叶绿体连续片段时,一些完整路径的类叶绿体连续片段将从组装图中移除。基于假设,即线粒体基因组的拓扑应表示为单一圆形或线性分子【26】,PMAT使用assembly_graph.py移除组装图中的一些尖端连续片段。尖端连续片段被定义为那些既不连接到组装图中的任何其他连续片段也不以圆形连接到自己的连续片段【26】。此外,如果一条路径的深度小于其两端连接的连续片段的五分之一,则将从组装图中移除该路径。PMAT还为用户提供了—minLink选项,以便在autoMito和graphBuild模式中直接移除假路径。为了补偿简化过程中可能的不足,PMAT为用户提供了GFA格式的原始组装图,以便在Bandage【66】中手动解开。
步骤6. 导出所有可能的构型和手动完成
然后,PMAT使用简化的组装图(PMAT_master.gfa)和连续片段标签信息(PMATContigGraph.txt)进一步导出所有可能的构型(s)到GFA和FASTA文件(s)。首先,PMAT将所有类线粒体连续片段(PMAT_master.gfa)作为查询,并对照保守PCG数据库进行BLASTn搜索。接下来,按深度对所有BLASTn命中的连续片段进行排序,以计算它们的中位数,进一步用于移除“噪声”和非目标连续片段。然后,PMAT选择最大的单拷贝连续片段作为起点,以穷尽搜索所有可能的路径。单拷贝和多拷贝连续片段由它们的深度和连接确定。最后,每个线粒体基因组构型将被导出为一个独立的FASTA文件。当主图无法作为圆形路径解决或太复杂(许多重复)以至于无法解决时,PMAT将保守地导出最终构型。此时,建议使用Bandage【66】可视化简化的组装图(PMAT_master.gfa)并手动移除噪声和非目标连续片段。在移除完整路径的叶绿体连续片段和尖端连续片段后,可以根据每个连续片段的拷贝数手动解开修订后的组装图。使用Bandage合并所有可能的节点后,所有可能的独特路径(s)可以导出为FASTA文件(s)。每条路径代表目标线粒体基因组的一种可能构型。
线粒体基因组验证和注释
为了验证四个公开可用线粒体基因组的组装,它们的参考线粒体基因组从NCBI核酸数据库下载。如表1所示,下载的参考文献包括P. nutans(NC_046778.1)、A. thaliana Col-0(NC_037304.1)、C. europaea(BK059238)、H. annuus ANN1372-3(NC_023337.1)和M. domestica Costard(NC_018554.1)。所有参与组装的线粒体连续片段使用MacVector v18.2.5(MacVector Home)的Align to Reference模块映射到它们相应的参考线粒体基因组上。一些具有双重或三重测序深度的重复连续片段在对齐到参考线粒体基因组前被复制。如果组装的线粒体基因组覆盖了>95%的公开参考线粒体基因组,则认为是完整的。
尽管PMAT可以从WGS数据生成完整的线粒体基因组而无需分离mtDNA,但它可能会丢失一些关键的线粒体基因组信息。为了进一步验证PMAT的组装准确性,我们从M. domestica和P. trichocarpa的愈伤组织中分离纯化mtDNA,并在Illumina NovaSeq6000平台上进行测序。基于它们纯化mtDNA的测序数据,使用GSAT组装了M. domestica和P. trichocarpa mtDNA的线粒体基因组【38】。
其他未发布的线粒体基因组使用GeSeq【67】和MITOFY【63】进行注释。通过参考其他进化上相似的植物线粒体基因组使用BLASTN【65】手动检查和调整假定的PCGs。tRNA和rRNA基因使用tRNAscan-SE v1.21【68】确认。PCGs、tRNA基因和rRNA基因的注释使用MacVector v18.2.5整合。
评估植物线粒体基因组组装所需的最小测序数据
在进行大规模WGS项目之前,有必要评估组装线粒体基因组所需的最小测序数据。为了确定植物线粒体基因组组装所需的最小测序数据,我们使用Seqtk的‘sample’模块(https://github.com/lh3/seqtk)随机抽取总测序数据的一部分(默认:200 Mb、500 Mb和1 Gb)。对于基因组较大的物种,默认的抽样数据不足。因此,我们为H. annuus(基因组大小3.01 Gb)线粒体基因组组装随机抽取了1、2和3 Gb的数据,为T. chinensis(基因组大小10.24 Gb)线粒体基因组组装随机抽取了5和10 Gb的数据。表2显示了子抽样测序数据和估计的线粒体基因组测序深度。当组装图圆化并覆盖整个参考线粒体基因组时,在定义的测序深度下认为线粒体基因组是完整的。否则,需要重新抽样更多的测序数据进行组装。


![[网鼎杯 2020 朱雀组]Nmap1](https://img-blog.csdnimg.cn/direct/7e0b4a08882e4c2b9d516560e0bdc7fb.png)