零基础入门转录组数据分析——DESeq2差异分析
目录
- 零基础入门转录组数据分析——DESeq2差异分析
- 1. 转录组分析基础知识
- 2. DESeq2差异分析(Rstudio)
- 3. 结语
1. 转录组分析基础知识
1.1 什么是转录组?
转录组(transcriptome) 是指在某一生理条件下,细胞内所有转录产物的集合,这些产物主要包括信使RNA(mRNA)、核糖体RNA(rRNA)、转运RNA(tRNA)以及非编码RNA(ncRNA)。其中,编码RNA可以通过转录和翻译过程产生蛋白质,从而直接调控生命活动;而非编码RNA具有调节基因表达的作用,它们可以对染色体结构、RNA加工修饰及稳定性、转录和翻译、甚至蛋白质的转运及稳定性产生影响。
(简单来说:转录组是指一个活细胞在特定时间和环境下,所能转录出来的所有RNA的总和。因此,转录组分析就是为了了解基因的表达调控机制和功能。)
1.2 差异分析是什么?为什么要做差异分析?
转录组差异分析是一种生物信息学分析方法,旨在比较不同样本或条件下转录组数据的差异,以揭示基因表达的变化。
(简单来说:就是从上w个基因中找出不同样本中表达具有差异的基因,从而了解这些差异基因如何影响生物体的生理功能和疾病发生发展。)
1.3 在R中对于转录组数据都有哪些差异分析的方法?
包括但不局限于DESeq2、limma和edgeR等。
DESeq2: 基于负二项分布模型进行差异分析,考虑了测序深度和基因长度对count数据的影响,因此在处理高度变异的基因时表现较好。DESeq2还提供了多种标准化方法,可以帮助减少批次效应和其他技术噪声。
limma: 最初是为微阵列数据设计的,后来也扩展到RNA-Seq数据分析。它使用线性模型进行差异分析,并且可以轻松处理多个因素和协变量。limma的优势在于其速度和灵活性,可以处理大量的基因和样本。
edgeR: 也是基于负二项分布模型进行差异分析,但与DESeq2不同的是,它使用了经验贝叶斯方法来估计分散参数,从而提高了差异检验的准确性和稳定性。edgeR在处理大型转录组数据集时表现出色,尤其是在识别低表达水平的差异基因方面。
1.4 三种方法对于输入数据的要求?
DESeq2: 需要原始的count矩阵(如通过htseq-count工具获得),并且只支持重复样品。
limma: 可以接受原始的count矩阵,但需要用户自行进行标准化(通常是log转换),并且也支持重复样品。
edgeR: 要求输入原始的count矩阵,既支持单个样品也支持重复样品。
1.5 在做转录组分析的时候该选用哪种方法?(关键)
GEO芯片数据——用limma差异分析,因为GEO芯片数据底层已经对数据做了标准化处理。
(数据参考:零基础入门转录组分析——数据处理(GEO数据库——芯片数据))
GEO高通量数据——用DESeq2差异分析,因为可以获取到count数据。
(数据参考:零基础入门转录组分析——数据处理(GEO数据库——高通量测序数据))
TCGA_count数据——用DESeq2差异分析,因为可以获取到count数据。
(数据参考:零基础入门转录组分析——数据处理(TCGA数据库))
自测序数据——用DESeq2差异分析,因为可以获取到count数据。
(数据参考:零基础入门转录组分析——数据处理(自测序数据))
注:目前了解到的很少用到edgeR,一般来说有重复的样本通常limma和DESeq2就能解决(edgeR这种方法不做过多介绍)。
2. DESeq2差异分析(Rstudio)
本项目以GSE203346数据集(高通量测序数据)展示DESeq2差异分析过程
实验分组:疾病组(18例),对照组(19例)
R版本:4.2.2
R包:tidyverse,lance,DESeq2
注:只要是能获取到count,并且有重复分组,都能用DESeq2做差异分析
数据处理过程参考之前的教程:零基础入门转录组分析——数据处理(GEO数据库——高通量测序数据)
设置工作空间:
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/XX/XX/XX') # 设置工作路径
if(!dir.exists('./01_DEGs')){
dir.create('./01_DEGs')
} # 判断该工作路径下是否存在名为01_DEGs的文件夹,如果不存在则创建,如果存在则pass
setwd('./01_DEGs/') # 设置路径到刚才新建的01_DEGs下
加载包:
library(tidyverse)
library(lance)
library(DESeq2)
导入处理好的表达矩阵:
dat <- read.csv('../../00_rawdata/GSE203346/dat.GSE203346_count.csv', check.names = F, row.names = 1)%>%lc.tableToNum()
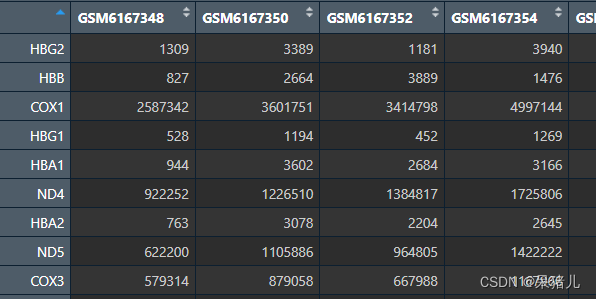
dat如下图所示行名为基因symbol,列名为样本ID,表中数字是原始count(都是整数,并且不需要做任何处理)
导入处理好的分组信息表:
# 分组信息
colData <- read.csv('../../00_rawdata/GSE203346/group.GSE203346.csv')
colData$group <- factor(colData$group, levels = c("control","disease"))
table(colData$group)
dat <- dat[, colData$sample] # 让表达矩阵的样本与分析信息表保持一致
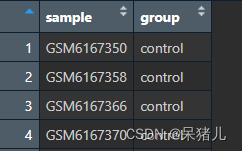
colData如下图所示,第一列为样本ID,第二列为样本分组
先创建DESeqDataSet对象(这里会用到前面导入的 dat和colData )
dds <- DESeqDataSetFromMatrix(countData = dat, colData = colData, design = ~group)
dds如下图所示
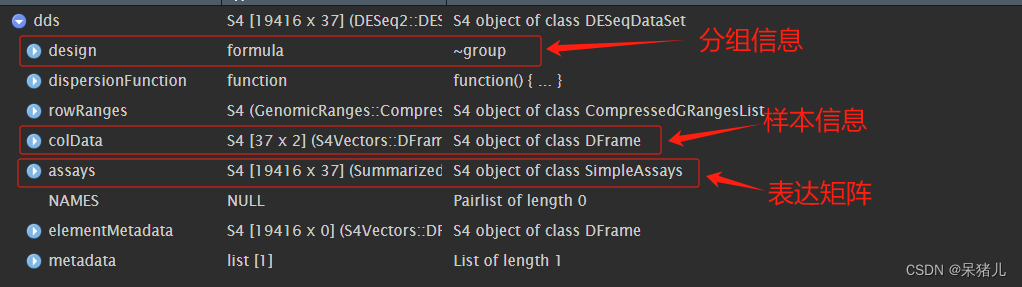
- design = ~group:这定义了实验的分组情况
- colData = colData:这是一个数据框,包含样本的列信息(例如,样本名、组别等)。
- assays = dat:count表达矩阵,行代表基因,列代表样本。
先筛选掉低表达的基因(为了尽量避免低表达基因导致的假阳性结果),之后估计每个样本的大小因子(大小因子是用于校正不同样本之间测序深度差异的一种方法,以确保在进行基因表达比较时能够公平地比较不同样本)。
dds <- dds[rownames(counts(dds)) > 1, ] # 过滤掉在任何样本中计数都小于或等于1的基因。
dds <- estimateSizeFactors(dds) # 估计每个样本的大小因子
estimateSizeFactors函数会根据每个样本的测序数据(通常是基因表达计数数据)来计算一个系数,这个系数反映了该样本相对于其他样本的测序深度,这个系数就是大小因子。
具体来说,DESeq2首先会对原始的表达量矩阵进行log转换,然后计算每个基因在所有样本中的均值。接下来,对于每个样本,它会将该样本中每个基因的表达量减去对应的所有样本中的均值,然后取中位数。最后,通过对这些中位数进行指数运算,得到每个样本的大小因子。
一旦计算出了每个样本的大小因子,就可以将其用于归一化原始的表达量数据。
归一化的目的:是消除不同样本之间由于测序深度差异而导致的偏差,使得不同样本之间的基因表达数据可以更加公平地进行比较和分析
之后通过DESeq函数用归一化后的数据进行差异表达分析
dds <- DESeq(dds)
最后提取差异表达分析的结果,并将其转换成数据框
res <- results(dds, contrast = c("group","control","disease"))
DEG <- as.data.frame(res)
注:results函数中contrast参数来指定比较时,第二个元素(在这个例子中是"control")通常是被用作参考组或基线组,而第三个元素(在这个例子中是"disease")则是用来和参考组进行比较的组。(换句话说,"control"是被比较的组,而"disease"是用来比较的组)
举个栗子:如果某个基因在"disease"中的表达量高于在"control"中的表达量,那么它的log2倍数变化将是一个正值
DEG 如下图所示:
- baseMean: 这一列显示的是每个基因在所有样本中的平均表达量,经过大小因子校正后的值。它反映了基因的总体表达水平。
- log2FoldChange: 这一列展示的是基因表达的对数2倍变化值(log2 fold change),正值表示在disease中表达上调,负值表示在disease中表达下调。
- lfcSE: 这一列是log2FoldChange的标准误差估计值,它提供了log2FoldChange估计的不确定性度量
- stat: 这一列是基因表达变化的统计检验值,用于衡量基因表达变化的显著性。一个大的正值或负值表示基因表达的变化是否显著。
- pvalue: 这一列显示的是基因表达变化的p值,用于评估观察到的效应(如基因表达的变化)是否可能是由于随机误差引起的,通常认为P < 0.05则结果是显著的。
- padj: 这一列是调整后的p值,用于控制假阳性率(简单说就是矫正后的P值)
给差异分析结果添加上下调标签
DEG$change <- as.factor(
ifelse(DEG$pvalue < 0.05 & abs(DEG$log2FoldChange) >= 0.5,
ifelse(DEG$log2FoldChange > 0.5,'Up','Down'),'Not'))
table(DEG$change)
DEG_write <- cbind(symbol = rownames(DEG), DEG)
处理好的 DEG_write 如下图所示,除了新添加的symbol和change,其余与DEG一样,change列中: (筛选条件:|log2FoldChange| >= 0.5 & p.value < 0.05)
- Up 表示上调基因
- Down 表示下调基因
- Not 表示未通过筛选的基因
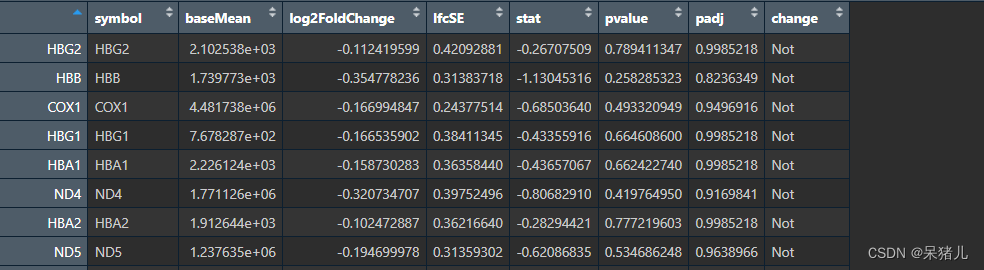
筛选出表达具有差异的基因 (筛选条件:|log2FoldChange| >= 0.5 & p.value < 0.05)
sig_diff <- subset(DEG, DEG$pvalue < 0.05 & abs(DEG$log2FoldChange) >= 0.5)
sig_diff_write <- cbind(symbol = rownames(sig_diff), sig_diff)
sig_diff_write 与DEG_write的差别就是少了那些没有差异的基因。
最后,保存差异分析的结果:
write.csv(DEG_write, file = paste0('DEG_all.csv'))
write.csv(sig_diff_write, file = paste0('DEG_sig.csv'))
3. 结语
以上就是DESeq2差异分析的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。
如果觉得本教程对你有所帮助,点赞关注不迷路!!!
- 目录部分跳转链接:零基础入门生信数据分析——导读