GEO数据挖掘
-
数据库:GEO、NHANCE、TCGA、ICGC、CCLE、SEER等
-
数据类型:基因表达芯片、转录组、单细胞、突变、甲基化、拷贝数变异等等
-
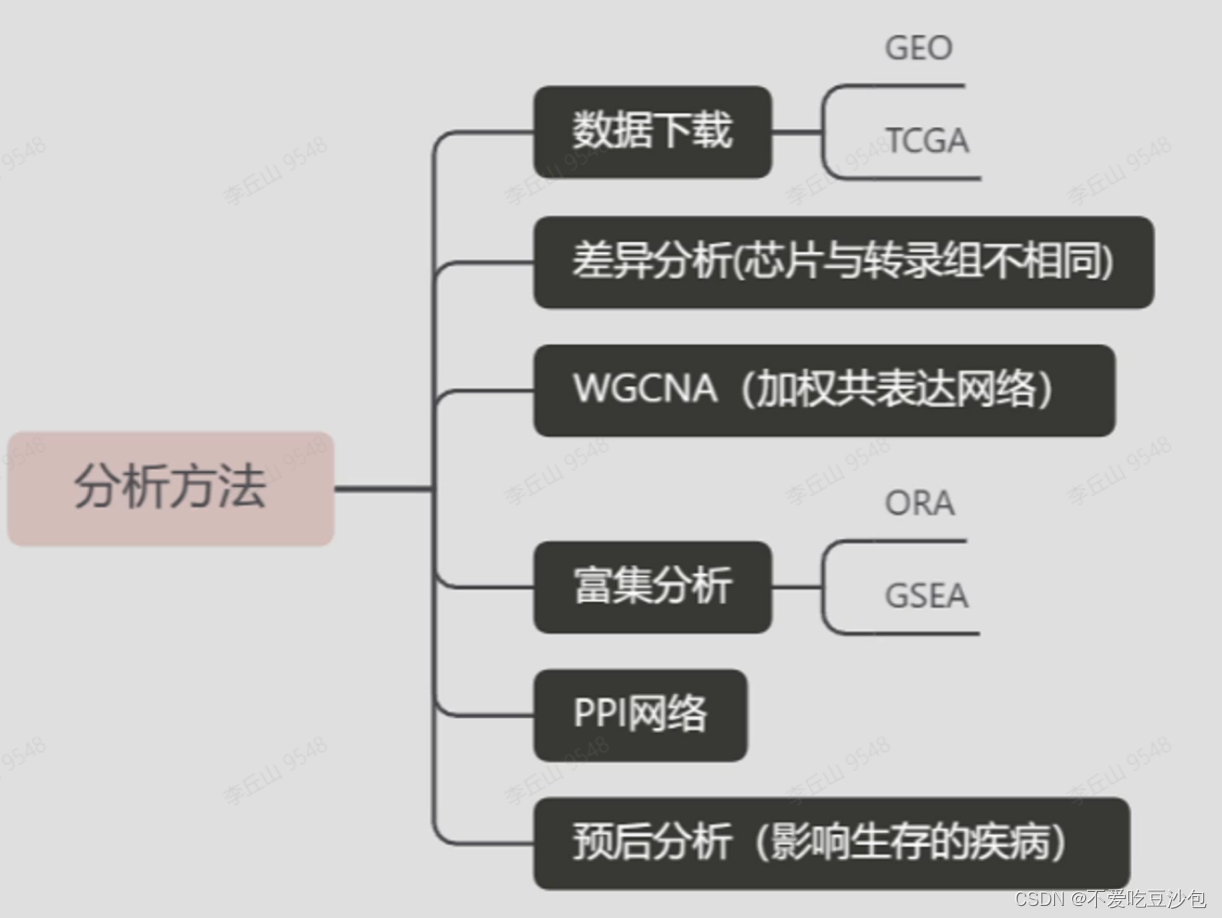
常见图表
表达矩阵
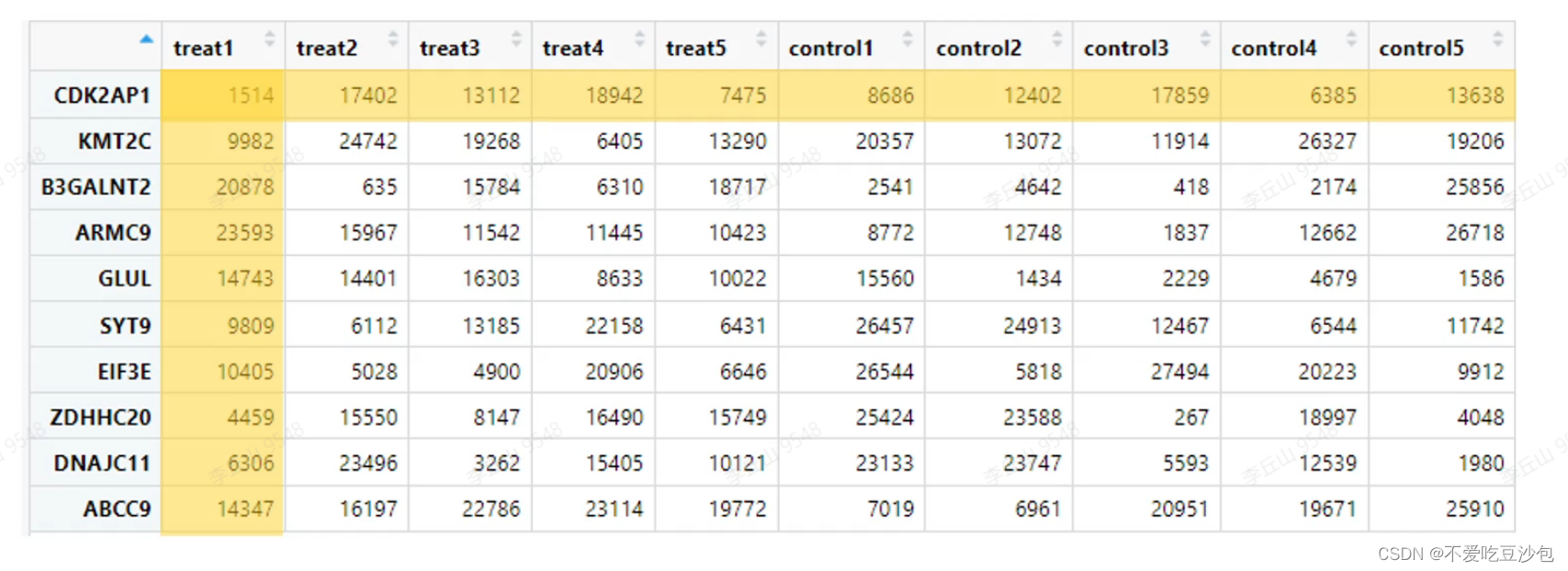
- 一行为一个基因,一列为一个样本,内容是基因表达量。
热图
输入数据是数值型矩阵/数据框
颜色变化表示数值大小
- Complexheatmap:自行探索~可以定义某些需要标注的基因。
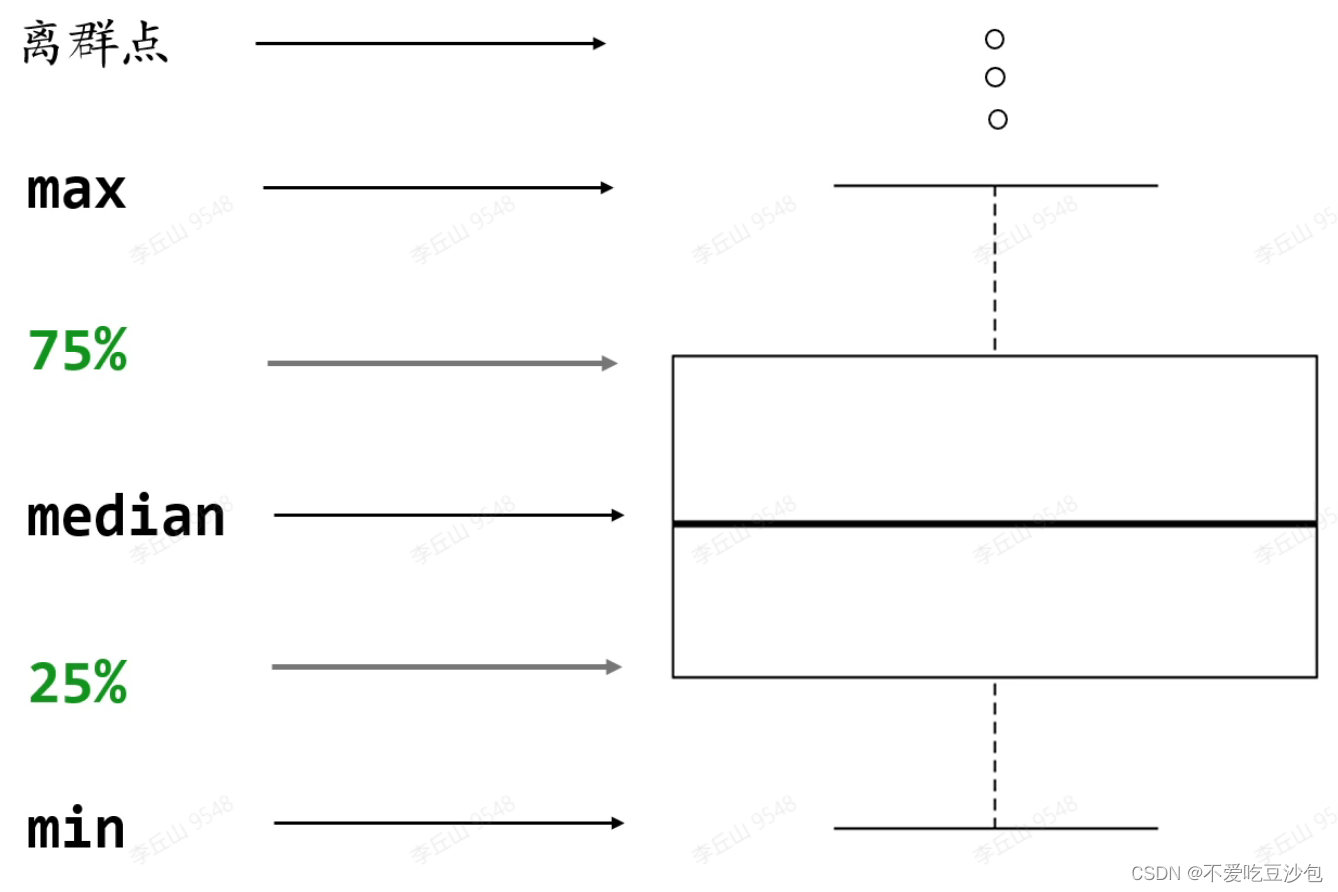
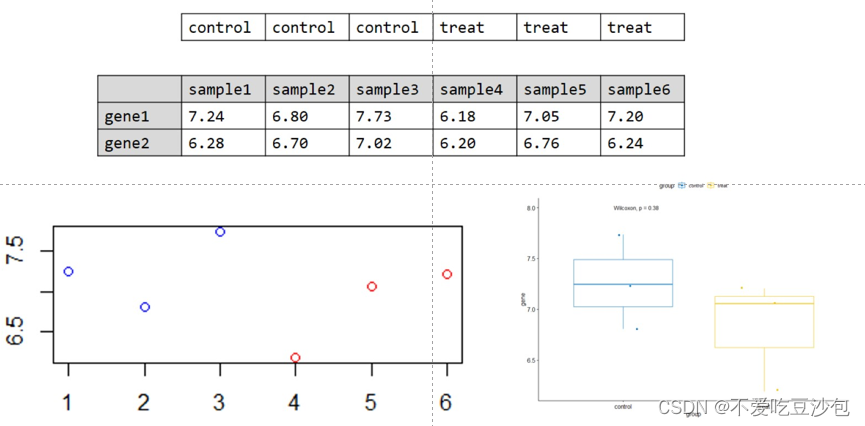
散点图和箱线图
-
数据要求:xy轴分别为一个连续型向量和一个有重复值的离散型向量(分类向量)
-
数据意义:箱线图会删除离群点
-
意义:单个基因在两组之间的表达量差异
矩阵/数据框中无法直接添加分组信息,因此需要一个单独向量来说明样本的分组信息。
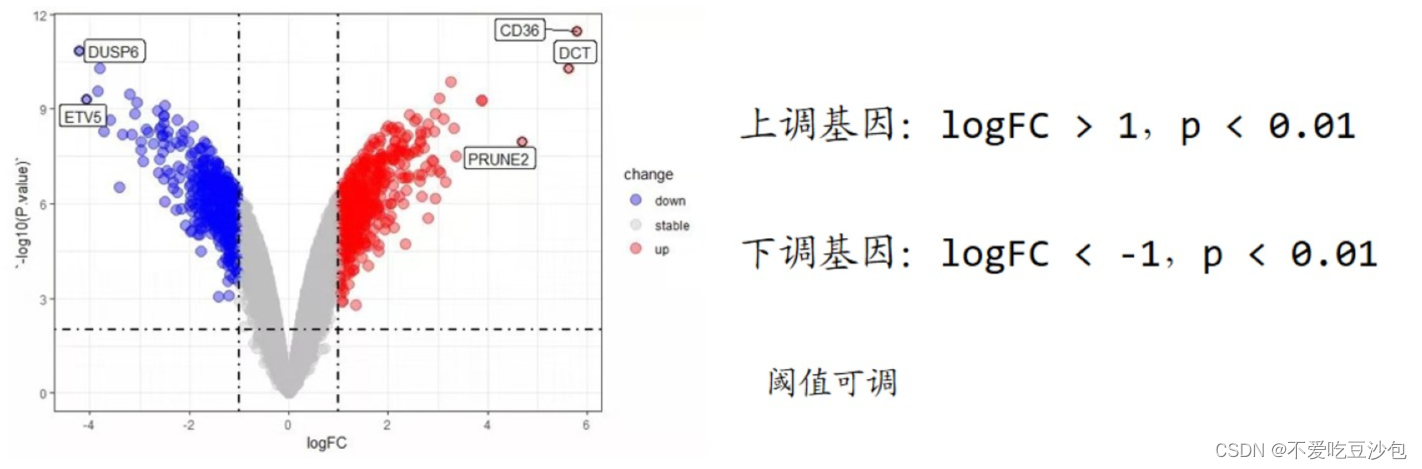
火山图
-
意义:展示多个基因在两组间的表达量差异
-
横坐标:FC(Foldchange) 处理组平均值/对照组平均值;logFC(log2Foldchange)
-
芯片差异分析的起点是一个取过log的表达矩阵,如果未取log需要自行log

-
-
理解logFC:log2(X/Y)=log2(x)-log2(y)
- log后的表达矩阵:表达量在0-24之间。
- 未log的表达矩阵,表达量在0,10,100,1000…
- logFC的正常范围:个位数居多。
-
logFC>0,treat>control,基因表达量上升;logFC<0,treat<control,基因表达量下降。通常说的上调和下调基因是指表达量显著上升下降的基因。
-
logFC常见的阈值:1/2/1.2/1.5/2.2/0.585[log2(1.5)];需要根据情况取值。
-
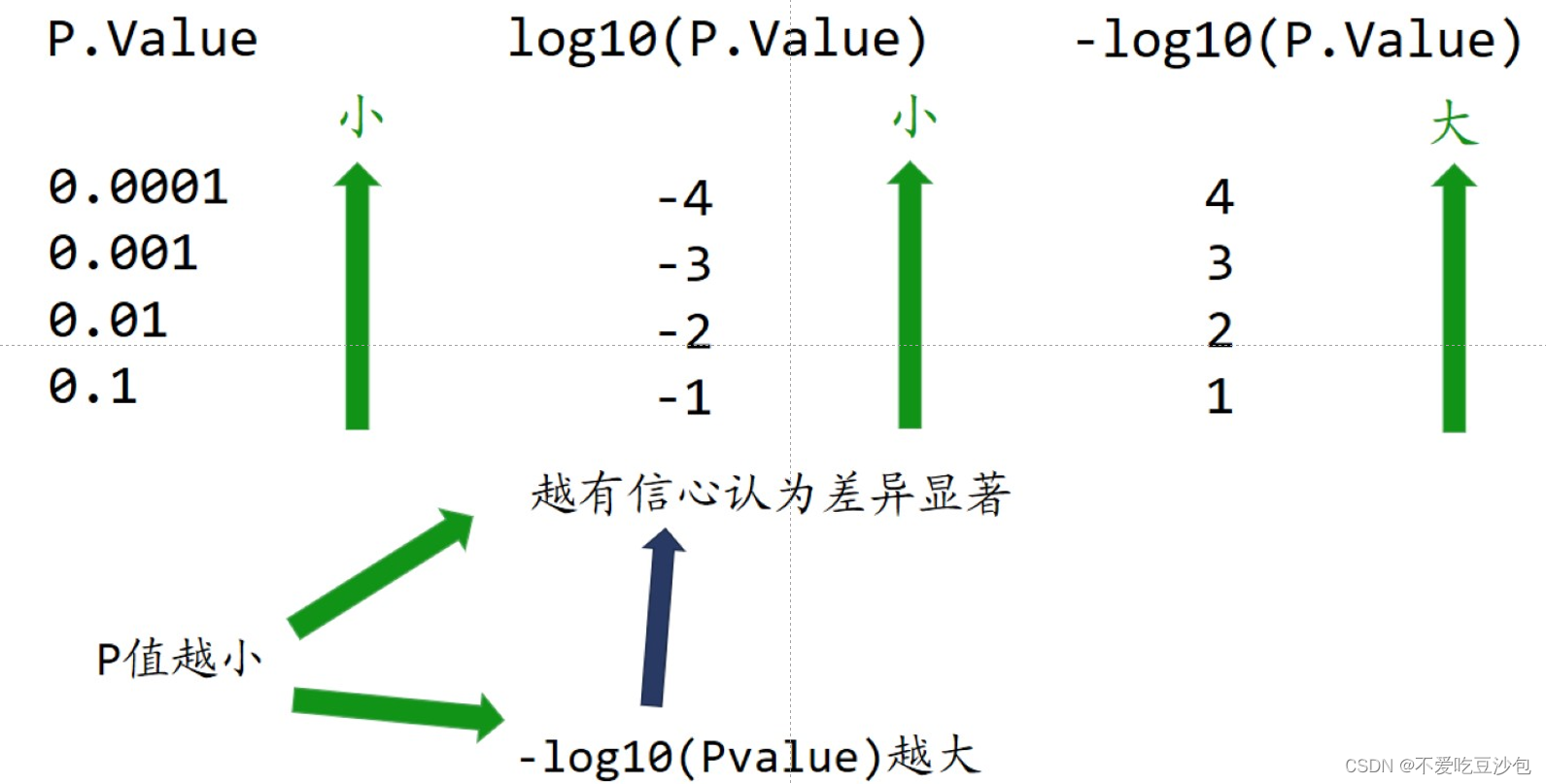
纵坐标:P.Value→log10(P.Value)→-log10(P.Value)
主成分分析
-
PCA:主成分,把多个指标转换成少数综合指标(主成分)来代表样本。代表样本的点在坐标轴上距离越远,说明样本差异越大。
-
PCA样本聚类图:dim1/dim2中数字不重要(尽量大)。
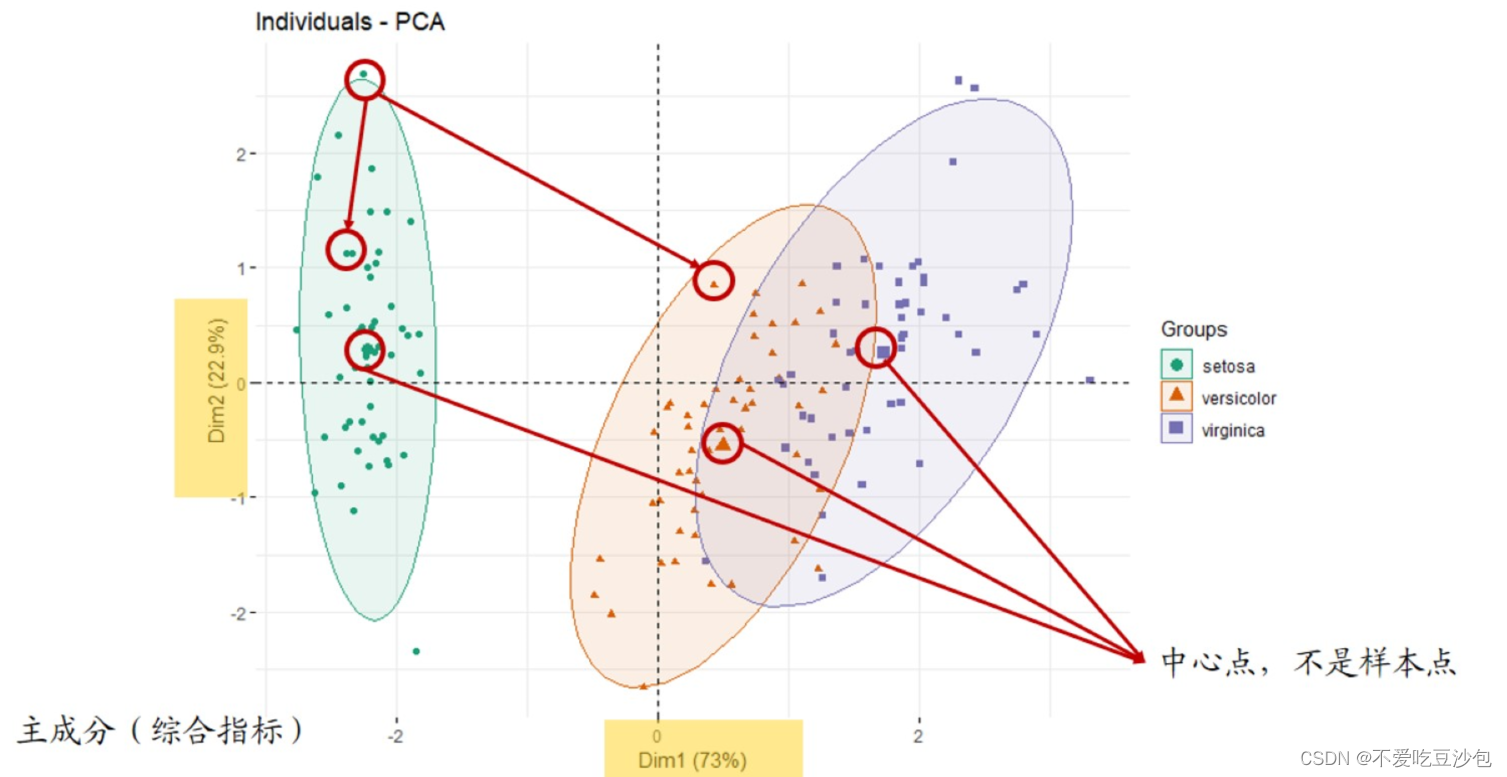
- 图上点代表样本(中心点除外),点与点之间距离代表样本差异。
- 可以用于“预实验”,简单看组间是否有差别。
表达芯片分析思路
表达数据实验设计
- 实验目的:通过基因表达量数据的差异分析和富集分析来解释生物学现象。
- 设计实验组和对照组
- 有差异的材料→差异基因→找功能→解释差异,缩小差异范围。
基因表达芯片
探针的表达量代表基因的表达量
探针根据要测量的基因设计,是一段与基因互补杂交的短核苷酸序列。探针和序列绑定,不和基因绑定。
数据库介绍
-
GEO数据库
- GEO工具:GEO2R,可以导入代码进入R修改。
-
Series:用户提交给数据库一个完整的研究,包括其样本数据(GSM),包含提供研究描述,包括对数据描述并总结分析(GSE)。
-
GEO数据集筛选
表达芯片数据:Expression profiling by array
单细胞/普通转录组(高通量测序):Expression profiling by high throughput sequencing
-
GSE界面:GPL(平台)中看ID和Symbol Gene;GSM(样本)中看表达量是否正常、是否需要取log等。
分析思路
- 找数据,找到GSE编号
- GEO数据库中检索
- 文献中查找GSE编号
- 下载数据:表达矩阵、临床信息(分组信息)、GPL编号(探针注释)
- 网页中点选下载
- 代码下载(推荐)
- 数据探索:分组之间是否有差异、PCA、热图(方差排名靠前的1k个基因)
- 差异分析和可视化:P值、logFC;火山图、热图
- 富集分析:KEGG、GO
表达矩阵
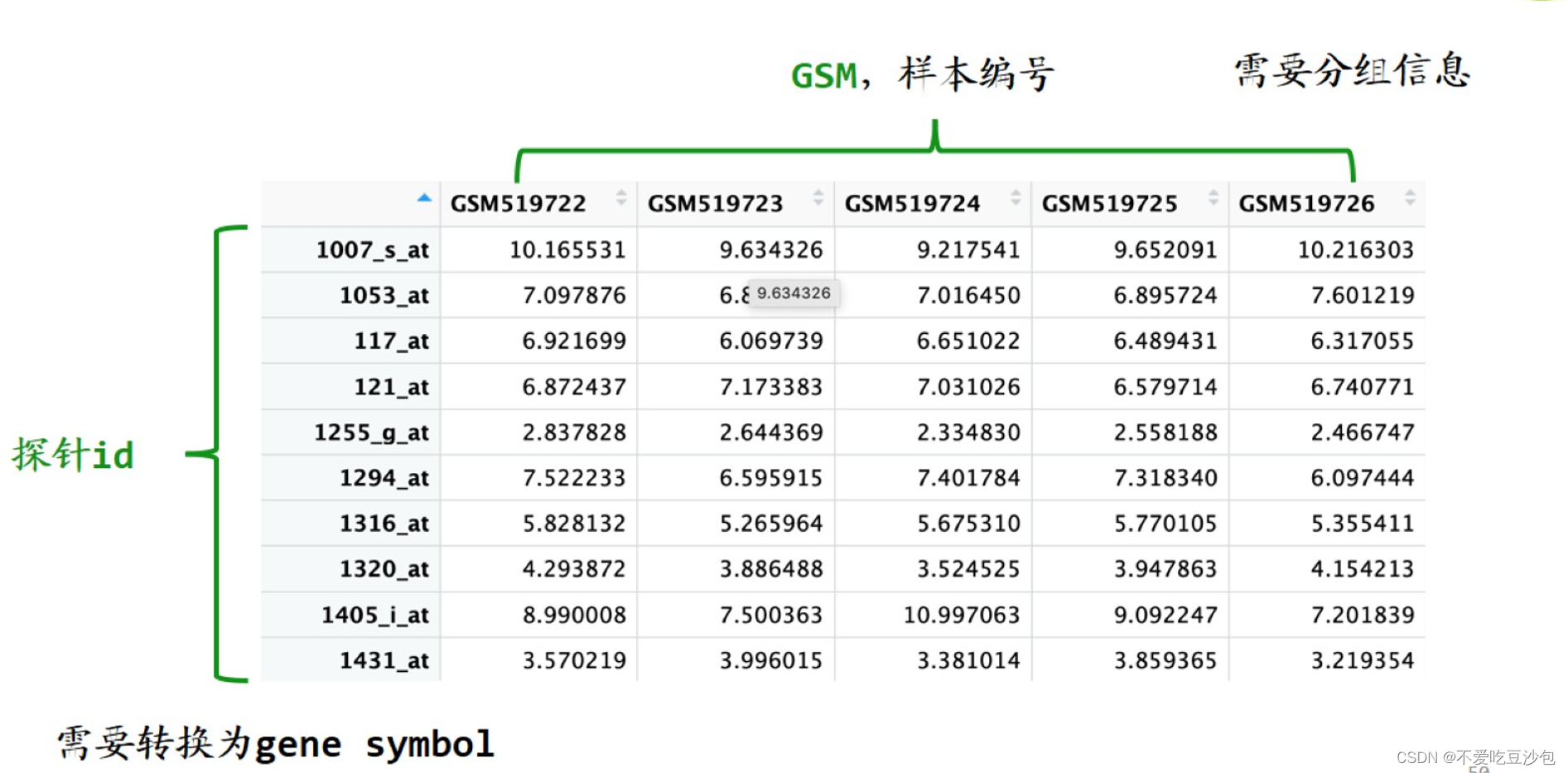
以分组为单位说问题,而不是以样本为单位
代码分析流程
安装R包
下载数据
library(GEOquery)
eSet = getGEO("GSE7305", destdir = '.', getGPL = F)
##探索eSet
class(eSet)
##[1] "list"
length(eSet)
##[1] 1
eSet = eSet[[1]] ##将list解开
class(eSet) ##是一种特殊的数据类型,可以从帮助文档中找到详细说明。出自Biobase包
##[1] "ExpressionSet"
##attr(,"package")
##[1] "Biobase"
提取表达矩阵
exp <- exprs(eSet) ##提取表达矩阵
dim(exp) ##查看数据属性
range(exp) ##查看数据范围,决定是否需要取log,是否有负值
##[1] 5.020951 22011.934000 ##这样的数据就需要取log
exp = log2(exp+1) ##取log
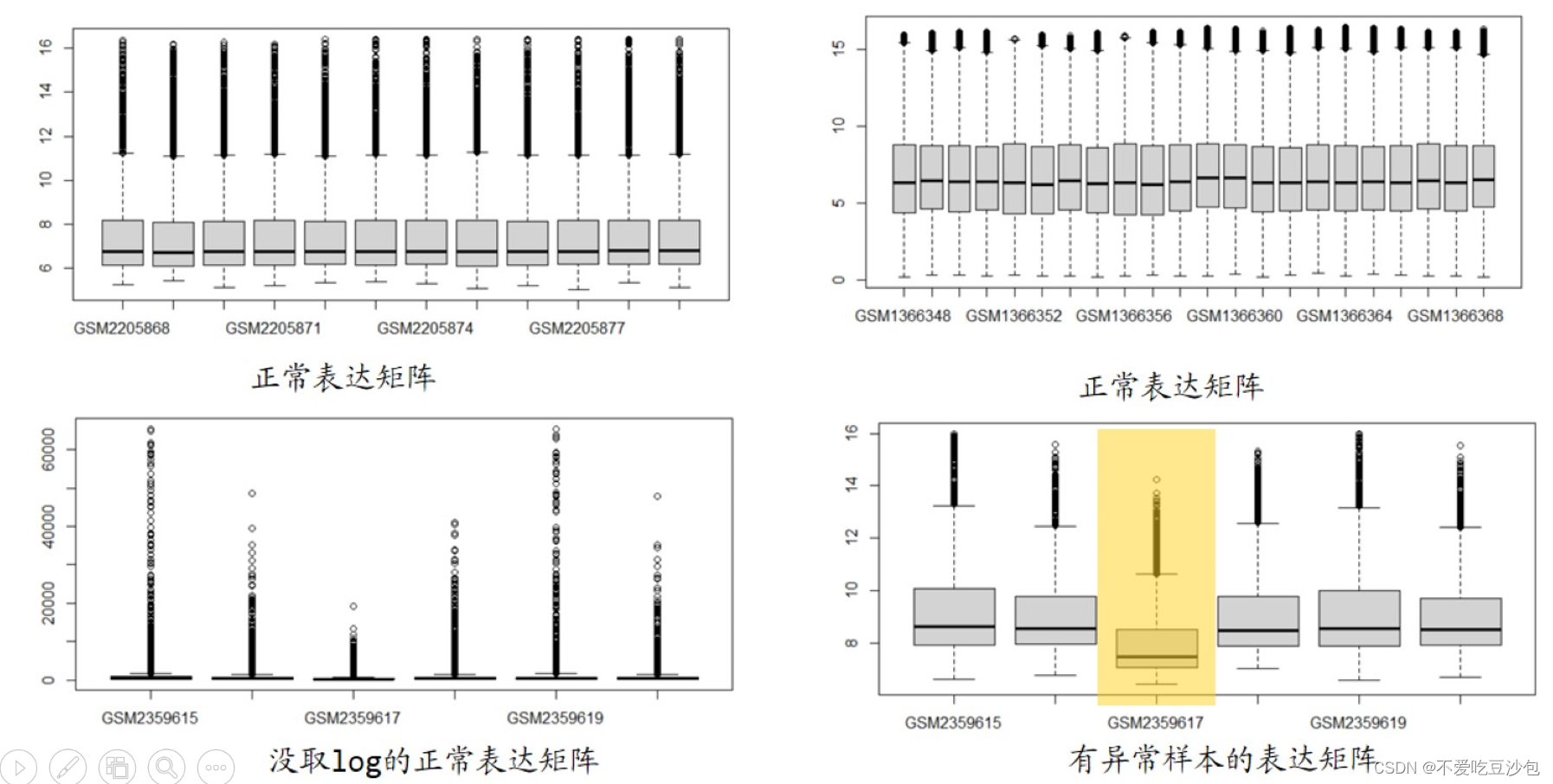
boxplot(exp,las = 2) ##检查数据情况
- 有异常样本:1. 删掉异常样本;2. limma包的标准化函数拉齐。
- 负值:log后少量负值可以接受,log前有负值或一半都是负值(标准化)弃用。
- 如果logFC在2-4之间,有可能取了2次log
提取临床信息
pd <- pData(eSet) ##包含分组信息
让表达矩阵和临床信息顺序匹配
p = identical(rownames(pd),colnames(exp));p ##判断是否对应
if(!p) { ##如果不对应,运行
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
}
提取芯片平台编号
gpl_number <- eSet@annotation;gpl_number ##等同于网页查找
save(pd,exp,gpl_number,file = "step1output.Rdata") ##保存数据
- 原始数据处理方法(有时间再整理吧~):https://mp.weixin.qq.com/s/0g8XkhXM3PndtPd-BUiVgw
引用自生信技能树课程~ 给小洁老师比心~