百趣代谢组学文献分享,SWATHtoMRM: Development of High-Coverage Targeted Metabolomics Method Using SWATH Technology for Biomarker Discovery,是由中国科学院生物与化学交叉研究中心,Dr. Zheng-Jiang Zhu课题组发表在AC上的一篇技术型文章。
代谢组学文献分享—摘要
生物样本代谢组存在高度复杂性,对大规模定量研究这些代谢物带来极大的挑战,严重限制了代谢组学在生物标志物发现研究中的运用。靶向代谢组学研究主要运用MRM技术,具有极高的定量能力,但却受制于较为有限的覆盖度。为了解决这一挑战,作者开发了一项新的技术策略——SWATHtoMRM,巧妙的运用了SWATH技术的高覆盖度特点来开发高覆盖度的靶向代谢组学方法。
首先,运用SWATH技术对一个生物混样进行非靶向代谢谱分析,以获取其中“所有”代谢物的二级谱图 (MS²);然后从中提取MRM离子对信息,用于构建高达1000-2000代谢物覆盖度的靶向代谢组学分析;随后,作者又展示了这种方法在定量代谢组学中优势,例如:覆盖度、重复性、灵敏度和动态范围等。最后,作者运用SWATHtoMRM技术寻找colorectal cancer (CRC)诊断标志物,单针进样便可完成多达1303种代谢物MRM检测。从CRC病人组织中发现并验证了20种潜在诊断标志物。其中17种标志代谢物在CRC病人血浆中被进一步验证与肿瘤切除相关,有着极大的CRC术后预后标志物潜力。
SWATHtoMRM技术为我们提供了一种全新的途径来开发高覆盖度的靶向代谢组学方法,将促进靶向代谢组学在疾病标志物发现研究中的应用。
代谢组学文献分享—背景
代谢组学旨在系统性定量检测所有代谢物的动态变化,为疾病表型提供一个综合的特征描述,以便于疾病标志物的发现。代谢物具有广泛的结构多样性和悬殊的浓度范围(>109)。因此,代谢组学研究要求分析技术具有广泛覆盖度、高灵敏性和特异性以及宽动态范围。
液质联用(LC-MS)技术是较为流行的代谢谱分析技术之一,包括靶向和非靶向两种方式。非靶向代谢组学多运用高分辨质谱(TOF/Orbitrap)来检测尽量多的代谢物,具有很宽的覆盖度,但是受限于动态范围、定量精度以及灵敏度等。相反,基于LC-MS的靶向代谢组学技术主要使用MRM技术,能够精确定量一定数量(100-300)的已知代谢物。
靶向代谢组学技术作为代谢物定量的金标准,具有较高的灵敏度、动态范围和较好的重现性。然而,较低的代谢物覆盖度成为了靶向代谢组学的主要运用瓶颈,因此,开发一种具有高覆盖度的靶向方法来准确定量在非靶向代谢组学中检测到的已知/未知代谢物至关重要。
针对MRM覆盖度低这一瓶颈,近期已有多篇文章着手开发高覆盖度的靶向代谢组学技术。 但这些技术往往需要很长的机时(20-30针, 有的甚至需要80针)来获取代谢物的二级谱图,严重限制了该技术的通量;使用常规的DDA采集模式来获取代谢物二级谱图也是这些方法中的一种,但由于DDA模式本身的随机性和偏好性,使得这种采集方式重复性低,覆盖度也偏低。不同于DDA采集模式,DIA/SWATH模式能够高效、高覆盖度获取“所有”代谢物的二级谱图,在近几年被不断开发用于非靶向代谢组学研究。
尽管SWATH技术能够同时采集所有代谢物的二级碎片离子信息,但是在复杂生物样品中,它的定量动态范围和重复性都受到了很大的限制。正是由于任何单一技术都不是广泛适用的,本文作者集各家之所长,将SWATH技术强大的定性能力与MRM技术的精确定量能力完美结合,开发出了SWATHtoMRM技术。
代谢组学文献分享—方法
LC-MS方法
为了保证SWATH采集到的代谢物二级碎片离子信息与MRM中所使用的Q1/Q3离子对信息相对应,需要保证液相条件(RT)完全一致,并尽量保证SWATH采集所使用的Q-TOF质谱仪(TripleTOF 6600)与MRM采集所使用的QQQ质谱仪(Agilent 6495 QQQ)具有相对一致的各项参数(CE,DP等)。
数据处理方法
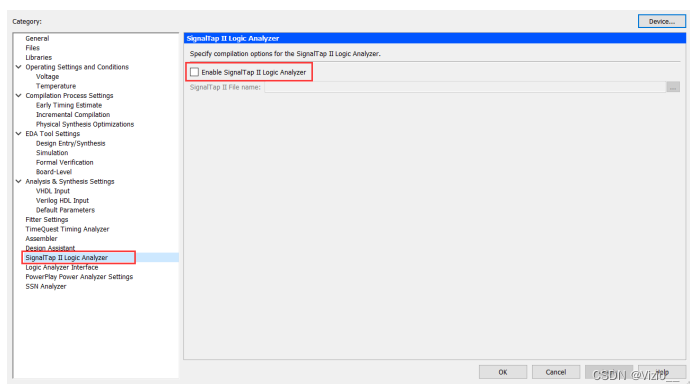
SWATHtoMRM流程
SWATHtoMRM技术的分析流程已整理成R包,用于从SWATH数据中大规模提取MRM离子对信息,该R包可以在(http://www.zhulab.cn/software.php)下载安装。整个流程分为两步(图1):
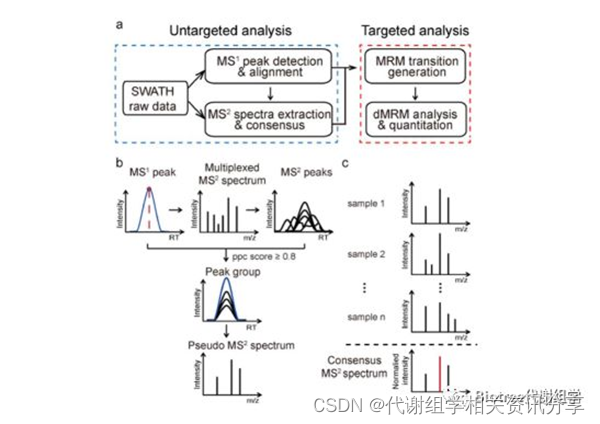
图1. (a) SWATHtoMRM 技术流程概览。 (b) 从SWATH数据中提取虚拟MS²图谱。 (c) 将来自多个样品的MS²谱图合并为consensus 谱图,保留出现频率超过50%的碎片离子,并用于生成MRM离子对。
1.非靶向分析SWATH数据
a) 使用XCMS包中的CentWave算法实现色谱峰(LC peak)识别,然后用CAMERA进行质谱峰(MS peak)注释,并去除同位素峰和小于1000的弱信号峰。对于多个数据文件,使用OBI-Warp算法进行峰对齐。→MS¹ LC peak 。
b) 针对每一个MS¹peak,从峰顶位置找到对应RT的混合MS²谱图,并过滤掉小于200的弱信号值、同位素离子以及大于母离子m/z的碎片离子。提取保留的碎片离子色谱峰(EIC)。→MS² LC peak 。
c) 通过计算每一个碎片离子EIC与母离子EIC之间的相关性得分(PPC score),将得分大于0.8的碎片离子EIC和对应母离子EIC归为一组,得到peak group(一个母离子EIC +多个碎片离子EIC),然后对每个peak group生成一张虚拟二级谱图。→ Pseudo MS² spectrum。
d) 将来自多个样品的相同MS¹ peak的二级谱图合并为一张谱图(consensus MS²)。合并原则为:保留出现频率超过50%的碎片离子,合并后的m/z为mean(m/z),intensity %为 mean(intensity %)。→ Consensus MS² spectrum。
2.生成MRM离子对信息
a) 采用3个原则评估consensus MS²中每一个碎片离子:(1)m/z(product ion)<m/z(precursor ion)-14.0126 Da,即在离子碎裂过程中至少有一个(CH₂)基团从母离子丢失;(2)剔除中性丢失H₂O、NH₃或者CO₂产生的碎片离子(也可自定义其他类型);(3)选择剩余例子中强度最高的碎片离子作为MRM离子对。最后,使用Agilent MassHunter构建schedule MRM方法,最小dwell 时间 5ms,cycle time为990ms。
代谢组学文献分享—结果与讨论
SWATHtoMRM 流程评估
首先,作者测试了SWATHtoMRM技术在多种生物样品,例如,人类尿液、结肠直肠组织以及Jurkat细胞中的适用性。
以人尿液样品为例,3554个MS¹ feature在SWATH数据中被检测到,其中950个同位素峰和弱信号峰被初步过滤。剩余的2604个代谢物峰中,2091 (80.3%) 个代谢物具有合适的MRM离子对信息,其他代谢物峰由于不满足PPC > 0.8这一条件被进一步剔除。通过手动分析2091个MRM离子对结果发现,多达1614 (77.2%) 个代谢物成功在尿液样品中检测到(图2a)。
类似的,在其他类型样品中得到了接近的检出率(76 - 80%)。 本次实验中仅采用了正离子模式,但负离子模式也是完全适用的。
图2b详细演示了通过SWATHtoMRM构建MRM离子对的过程。 以taurine这个代谢物(m/z = 126.0212 Da,RT= 518 s)为例:首先,包含多个代谢物碎片离子的多重二级谱图被提取出来。然后根据与母离子EIC之间的相关性PPC,保留得分大于0.8 的碎片离子。随后,将来自多个样品的同一代谢物二级谱图合并,得到一张包含13个碎片离子的consensus二级谱图。最后,从中选择响应强度最高的碎片离子(m/z = 44.0496 Da)构建taurine的MRM离子对(Q1/Q3, 126.0/44.0)。
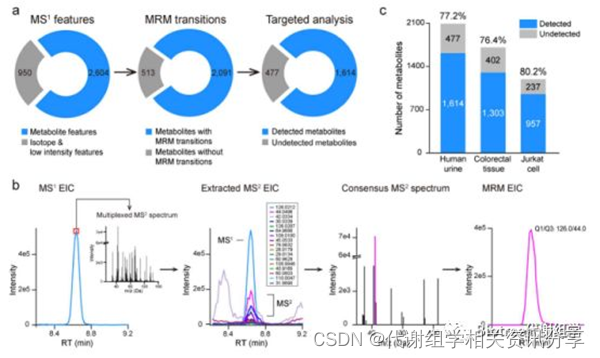
图2. (a) 使用人尿液样品大规模生成MRM离子对。(b) 代谢物taurine的离子对生成过程。 (c) 从不同类型生物样品(尿液,组织和细胞)中检测到的MRM离子对统计信息。
SWATHtoMRM技术的覆盖度评估
为了证实这一技术具有广泛的覆盖度,作者将SWATHtoMRM方法与DDA方法做了系统的比较。使用相同的尿液样品,相同的仪器参数,连续采集SWATH和DDA数据。后续的数据分析也采用相似的参数自动化进行,以较大的降低主观偏好性。从相同的尿液样品中,分别检测出2604(SWATH)和2149(DDA)个feature(图3a, 3b)。比较两者的二级谱图覆盖度,SWATH(2105, 80.8%)显著高于DDA(1174, 54.6%)。二级覆盖度在两者共享feature中,SWATH(84.9%)也显著高于DDA(61.1%)。
SWATH与DDA技术分别成功构建2091和1163个MRM离子对,其中852个是共享离子对(图3c)。在这852个共享离子对中,分别有87.2%(SWATH)和88.7%(DDA)的检出率,两种方法具有接近的检出率,说明SWATH技术生成的MRM离子对信息是可靠的(图3d)。 但是从总检出数上来看,SWATH技术比DDA多出66%的代谢物。简言之,SWATH技术具有与DDA相似的数据质量,同时又显著超越了DDA的检测数量。
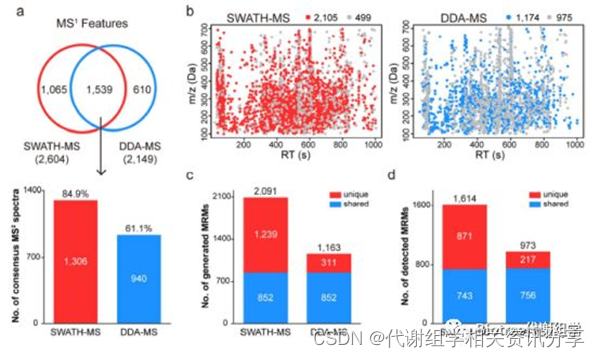
图3. 从SWATH数据中生成的MRM离子对具有很广的覆盖度。(a) 韦恩图展示了SWATH与DDA技术检测到的共享和特有feature。(b) SWATH与DDA数据中MS¹和MS²的分布图。红/蓝=有MS²,灰色=无MS²。(c) 两种方法构建的MRM离子对数目比较。(d) 两种方法检测到的代谢物数目比较。
SWATHtoMRM定量性能评估
为了评估SWATHtoMRM技术的定量性能,作者对比了SWATHtoMRM,SWATH-MS¹以及SWATH-MS² 3种技术间的灵敏度、动态范围和重复性。首先,梯度稀释尿液样品,分别进行SWATHtoMRM和SWATH采集,然后随机选择629个检测到的代谢物进行比较。
1.灵敏度:比较不同稀释梯度中代谢物检出数来评估,结果SWATHtoMRM > SWATH-MS¹= SWATH-MS²(图4a)。将这一比较细化到不同丰度范围代谢物中,同样是SWATHtoMRM优胜,尤其是在低丰度代谢物中(图4b)。
2.动态范围:比较不同稀释梯度中,629个代谢物的定量线性范围(R²)来评估,结果SWATHtoMRM > SWATH-MS¹> SWATH-MS²,SWATHtoMRM数据中R²> 0.8的代谢物超过了80%,而SWATH-MS¹和SWATH-MS²则不到60%(图4c)。
3.重复性:通过比较相邻稀释梯度的代谢物检出强度比值来评估。统计629个代谢物在两组相邻稀释梯度间检出强度比值(4×:16×、16×:64×,log2(理论值)=2),结果表明SWATHtoMRM > SWATH-MS¹> SWATH-MS²,SWATHtoMRM技术在不同的稀释梯度组合中具有接近的中位值以及较窄的分布,同时SWATH-MS¹也是优于SWATH-MS²的,SWATH-MS²在16×:64×组合中,比值中位数已经严重偏离了理论值(4倍),并且具有很宽的分布范围(图4e)。
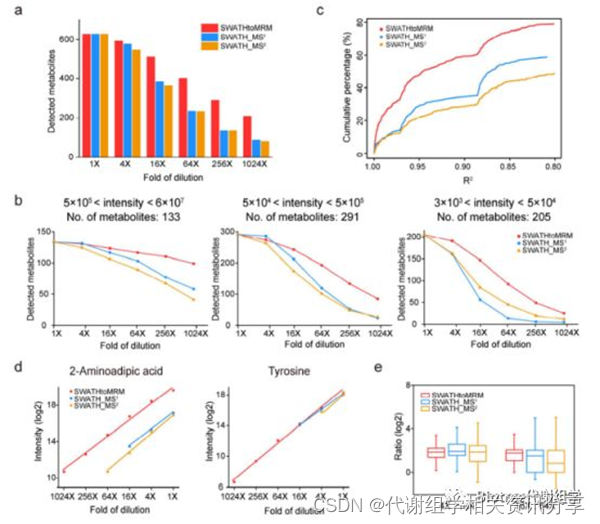
图4. SWATHtoMRM技术定量性能评估。(a) 3种方法在不同稀释梯度中的代谢物检出数比较。 (b) 细化到不同丰度范围时代谢物检出数。(c)R²累积分布,从R²=1到R²=0.8,满足条件的代谢物数目逐渐增多。(d) 列举了两种实际代谢物的定量线性范围。(e) 两个相邻稀释梯度间的比值分布图。
SWATHtoMRM实用性评估
为了评估SWATHtoMRM技术的实用性能,作者将该技术运用于结肠癌(CRC)诊断标志物的发现研究(18对样品作为训练集,42对用作验证集)。利用pooled QC样品构建了1705个MRM离子对,并成功检测到了其中的1303(76.4%)个代谢物。总共有1213(93.1%)个代谢物在>4个实际组织样品中加测到,并用于后续统计分析。从QC样品的RSD以及PCA分析结果来看,SWATHtoMRM在重复性和灵敏度方面显著优于SWATH-MS¹技术,主要表现在更低中位RSD值和较窄的RSD分布(图5a),以及PCA得分图中更小的组内离散度和更大的组间分离度(图5b)。
在生物标志物发现研究阶段,1303个代谢物中有358个具有统计学差异(fold-change >1.5, p-value > 0.01),其中67个被成功鉴定。随后,使用PLS-DA模型分析了CRC癌组织与癌旁组织的代谢物差异,VIP最高的20个代谢物被定义为潜在生物标志物(图5d),并具有极好的辨别能力(AUC = 1)。接下来,作者在验证集样品中检测了这20个代谢物,并评估了其预测精度,结果发现这些代谢物在验证集样品中同样具有极好的辨别能力(AUC=0.998,95% CI)(图5e)。
最后,作者使用CRC病人手术切除前后血浆样品来评估了这些代谢物的预后标志物潜力。其中17个代谢物表现出可靠的预测能力(AUC = 0.779,95% CI,sensitivity = 91.2%,specificity = 64.7%)(图5f)。
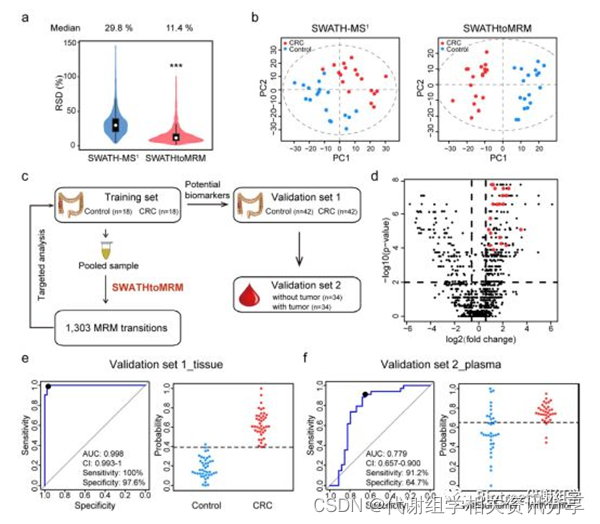
图5. (a) QC样品中检测到的1213个代谢物RSD值分布。(b) 分别使用SWATH-MS¹和SWATHtoMRM技术检测癌组织和癌旁组织中代谢物得到的PCA打分。(c)生物标志物发现研究的SWATHtoMRM实验流程。(d) 火山图展示1213个检测到的代谢物,红色点表示定义的20个潜在生物标志物。(e) 使用验证集组织样品的20个代谢物构建的PLS预测模型ROC曲线(左)和概率分布图(右)。(f) 使用验证集血浆样品的17个代谢物构建的PLS预测模型ROC曲线(左)和概率分布图(右)。
代谢组学文献分享—总结
作者开发了一种新的靶向代谢组学技术—SWATHtoMRM,可同时检测高达1000-2000个代谢物。并从多个角度详细对比了SWATHtoMRM与DDA、SWATH-MS¹以及SWATH-MS²技术之间的优劣。与DDA相比,SWATHtoMRM技术能够构建更多的MRM离子对。而与SWATH-MS技术相比,SWATHtoMRM具有更好的重复性、更高的灵敏度和更广的覆盖度。同时通过实际案例探究了该方法在代谢物生物标志物发现研究中的巨大潜力。