欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/134599076
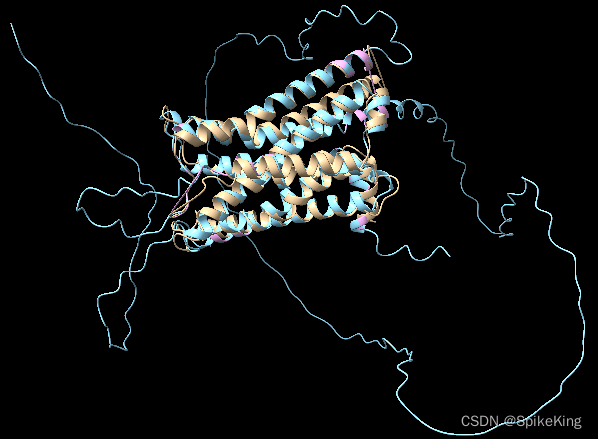
在蛋白质结构预测的过程中,输入一般是蛋白质序列(长序列),预测出 PDB 三维结构,再和 Ground Truth 的 PDB 进行比较,GT 的 PDB 是实验测出,比真实的蛋白质序列要短,需要使用算法进行查找。
满足约束:PDB 结构序列的长度 小于 蛋白质序列的长度,并且是子集关系。
- 黄色是 PDB 官网的结构
- 蓝色是预测的全长序列的结构
- 粉色是通过算法,截取的子结构
即:
源码:
def match_sub_seq(seq_long, seq_short):
"""
匹配序列子串,返回区间范围,一般用于长FASTA与短PDBSeq之间的预处理
"""
def get_seq_max_idx(seq_l, seq_s):
"""
序列匹配结果,返回连续最大索引
"""
np = len(seq_l)
nf = len(seq_s)
res = [0] * np
i, j = 0, 0
same = 0
is_next, next_i = True, 0
while i < np:
rp = seq_l[i] # pdb 的 残基
rf = seq_s[j] # fasta 的残基
if is_next:
next_i = i + 1
is_next = False
if rp == rf:
same += 1
j += 1
else:
j = 0
same = 0
if seq_l[i] == seq_s[j]:
same += 1
j += 1
if i < np:
res[i] = max(same, res[i])
i += 1
if j >= nf:
j = 0
same = 0
# 避免 "AAABCDEFGAB" 与 "AABCDEFG" 情况
if rp != rf:
i = next_i
is_next = True
return res
nl, ns = len(seq_long), len(seq_short)
size = 0
gap_list = [] # 区间范围
tmp_seq_short = seq_short
# print(f"[Info] seq_long: {seq_long}")
# print(f"[Info] seq_short: {seq_short}")
prev_idx = 0
while size < ns:
# print(f"[Info] tmp_seq_short: {tmp_seq_short}")
res = get_seq_max_idx(seq_long, tmp_seq_short)
max_val = max(res) # 最大索引值
max_indices = [i for i, x in enumerate(res) if x == max_val]
for j in sorted(max_indices):
# print(j, prev_idx)
if j <= prev_idx:
continue
else:
max_idx = j
break
s_idx, e_idx = max_idx-max_val+1, max_idx+1
prev_idx = e_idx
gap_list.append([s_idx, e_idx])
tmp_sub_long = seq_long[s_idx:e_idx]
tmp_short = tmp_seq_short[:max_val]
assert tmp_sub_long == tmp_short
tmp_seq_short = tmp_seq_short[max_val:]
size += max_val
# 验证逻辑
f_seq = ""
for gap in gap_list:
f_seq += seq_long[gap[0]:gap[1]]
assert f_seq == seq_short
return gap_list
从索引中,提取 PDB:
def extract_pdb_from_gap(pdb_path, output_path, gap_list):
"""
从残基的 gap list 提取新的 PDB 文件
"""
d3to1 = {'CYS': 'C', 'ASP': 'D', 'SER': 'S', 'GLN': 'Q', 'LYS': 'K',
'ILE': 'I', 'PRO': 'P', 'THR': 'T', 'PHE': 'F', 'ASN': 'N',
'GLY': 'G', 'HIS': 'H', 'LEU': 'L', 'ARG': 'R', 'TRP': 'W',
'ALA': 'A', 'VAL': 'V', 'GLU': 'E', 'TYR': 'Y', 'MET': 'M'}
d1to3 = invert_dict(d3to1)
# chain_idx = 0
atom_num_idx = 1
res_seq_num_idx = 0
pre_res_seq_num = "" # 残基可能是52A
chain_id_list = []
out_lines = []
line_idx = 0
lines = read_file(pdb_path)
res_ca_dict = dict()
for idx, line in enumerate(lines):
# 只处理核心行
record_type = str(line[:6].strip()) # 1~6
if record_type not in ["ATOM", "HETATM"]:
continue
record_type = "ATOM"
line_idx += 1
# 重新设置 atom_serial_num
# atom_num = int(line[6:11].strip()) # 7~11
atom_num = atom_num_idx
atom_num_idx += 1
# 替换为标准氨基酸
residue_name = str(line[17:20].strip()) # 18~20
if residue_name not in d3to1_ex:
continue
if residue_name in d3to1_ex and residue_name not in d3to1.keys():
a = d3to1_ex[residue_name]
residue_name = d1to3[a]
# 不修改链名
chain_id = str(line[21].strip()) # 22
if chain_id not in chain_id_list: # 更换链
chain_id_list.append(chain_id)
pre_res_seq_num = ""
# chain_idx += 1
# chain_id = chr(ord("A") + chain_idx - 1)
# 重新设置 res_seq_num
res_seq_num = line[22:27].strip() # 23~26 \ 23~27
if pre_res_seq_num != res_seq_num: # 更换残基
pre_res_seq_num = res_seq_num
res_seq_num_idx += 1
res_ca_dict[res_seq_num_idx] = False
res_seq_num = res_seq_num_idx
# 确保只有一个CA
atom_name = str(line[12:16].strip()) # 13~16
if atom_name == "CA":
if res_ca_dict[res_seq_num]:
print(f"[Warning] PDB res {res_seq_num} has more than one CA! ")
continue
else:
res_ca_dict[res_seq_num] = True
coordinates_x = str(line[30:38].strip()) # 31~38
coordinates_y = str(line[38:46].strip()) # 39~46
coordinates_z = str(line[46:54].strip()) # 47~54
occupancy = str(line[54:60].strip()) # 55~60
temperature_factor = str(line[60:66].strip()) # 61~66
element_symbol = str(line[76:78]) # 77~78
# 判断残基索引是否在 gap_list 中,其余保持不变
is_skip = True
for gap in gap_list:
if gap[0] <= res_seq_num-1 < gap[1]:
is_skip = False
if is_skip:
continue
out_line = "{:<6}{:>5} {:^4} {:<3} {:<1}{:>4} {:>8}{:>8}{:>8}{:>6}{:>6} {:>2}".format(
str(record_type), str(atom_num), str(atom_name), str(residue_name),
str(chain_id), str(res_seq_num),
str(coordinates_x), str(coordinates_y), str(coordinates_z),
str(occupancy), str(temperature_factor),
str(element_symbol)
)
out_lines.append(out_line)
create_empty_file(output_path)
write_list_to_file(output_path, out_lines)
def truncate_pdb_by_sub_seq(pdb_path, sub_seq, output_path):
"""
根据子序列提取新的 PDB 文件
"""
seq_str, n_chains, chain_dict = get_seq_from_pdb(pdb_path)
pdb_seq = list(chain_dict.values())[0]
try:
gap_list = match_sub_seq(pdb_seq, sub_seq)
except Exception as e:
print(f"[Warning] input sub_seq is not pdb sub seq! {pdb_path}")
return output_path
extract_pdb_from_gap(pdb_path, output_path, gap_list)
# 验证逻辑
seq_str, n_chains, chain_dict = get_seq_from_pdb(output_path)
new_seq = list(chain_dict.values())[0]
assert new_seq == sub_seq, print(f"[Error] new_seq: {new_seq}, sub_seq: {sub_seq}")
return output_path