今天给同学们分享一篇生信文章“Integrated PPI- and WGCNA-Retrieval of Hub Gene Signatures Shared Between Barrett's Esophagus and Esophageal Adenocarcinoma”,这篇文章发表在Front Pharmacol期刊上,影响因子为5.6。

结果解读:
选定研究的数据
作者从GEO和ArrayExpress数据库中收集了总共682个研究,截至2019年10月。其中,排除了678个不符合纳入标准的数据集/研究。最终选择了四个潜在的研究。在这四个选定的研究中,三个是在Affymetrix人类基因组U133A平台上进行的,一个是在Affymetrix人类基因组U133 plus 2.0平台上进行的,总共选择了125个样本。在每个研究中,EAC样本与相邻的BE样本进行了比较。使用数据集GSE37200构建了一个具有相关临床特征信息的共表达网络。经过预处理和归一化处理,进一步处理了包含22,284个基因的GSE37200数据集,并选择了变异基因(12,701个)进行WGCNA研究。
差异表达基因(DEGs)的鉴定和富集分析
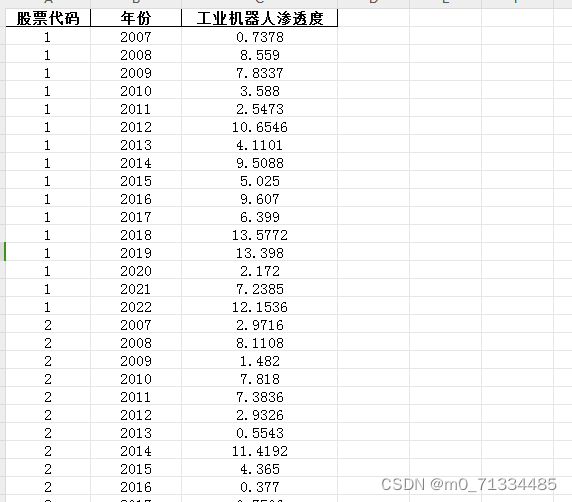
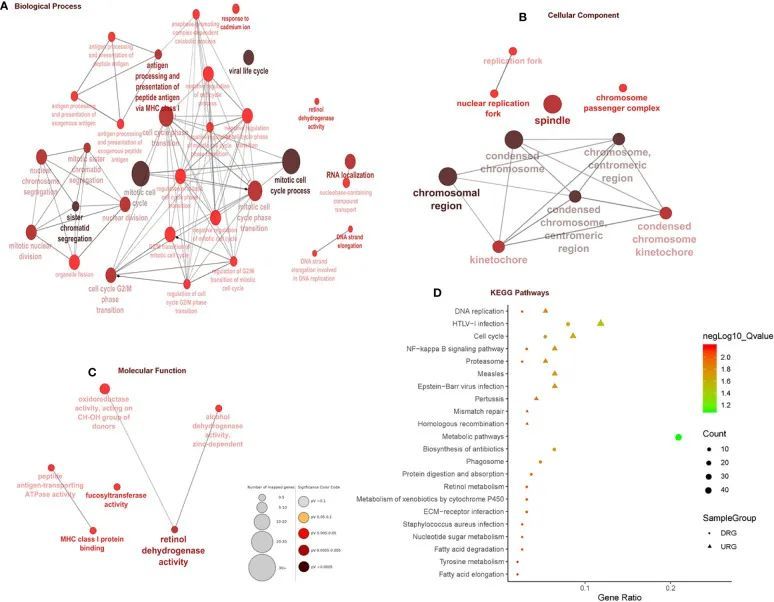
总共通过微阵列元分析获得了403个差异表达基因(DEGs),其中包括169个下调基因和234个上调基因。热图是一种简单而有效的比较多个主要基因列表内容的方法。热图中以红色、橙色和黄色表示所有数据集中的主要DEGs。灰色表示相应的基因在基因列表中不存在。表2展示了前10个上调和下调的DEGs。单核细胞分化抗原CD14(CD14)、核糖5-磷酸异构酶A(RPIA)、肿瘤坏死因子超家族成员11(TNFSF11)、plexin D1(PLXND1)、主要组织相容性复合体II DM beta(HLA-DMB)和剪接体相关因子3、U4/U6回收蛋白(SART3)是高表达的上调基因,而岩藻糖转移酶2(FUT2)、SECIS结合蛋白2类似物(SECISBP2L)、COP9信号体亚单位4(COPS4)、凝胶蛋白(GSN)和谷胱甘肽过氧化物酶3(GPX3)是高表达的下调基因。根据基因本体学(GO)术语BP、MF和CC,下调基因在有丝分裂细胞周期过程、姐妹染色单体分离、抗原处理、通过MHC I类呈递肽抗原、染色体区域和MHC I类蛋白结合等方面显著富集,而与EAC相关的上调基因中高度富集了视黄醇脱氢酶活性和岩藻糖转移酶活性(图2A-C)。在KEGG中,通路富集分析显示上调基因在病毒性心肌炎、细胞周期、DNA复制和糖基化终末产物-AGE-RAGE信号通路等方面富集。下调基因与脂肪酸降解、糖脂类代谢和氨基糖和核苷酸糖代谢等通路相关(图2D)。
WGCNA和临床相关模块鉴定
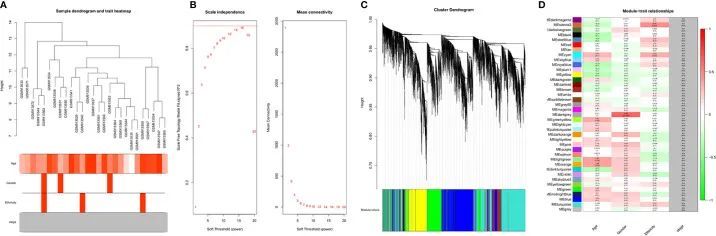
使用平均连接法和Pearson相关系数方法对带有临床特征的样本(GSE37200)进行了样本的树状图聚类(图3A)。进行了共表达分析以构建共表达网络。在本研究中,选择β=9(无标度R<0> = 0.95)作为软阈值参数,以确保无标度网络(图3B)。根据不相似度测量(1-TOM)对所有差异表达基因进行了树状图聚类(附图3)。通过层次聚类法确定了39个模块。浅绿色(特征基因值=0.41)、深灰色(特征基因值=0.62)和Sienna3(特征基因值=0.46)模块似乎与年龄、性别和种族有最高的关联。图3C中未显示与肿瘤分期相关的模块-特征关系,表示为NA。因此,选择与性别最相关的深灰色模块作为进一步分析的临床显著模块。在深灰色模块中确定了207个表型基因(图3D)。在补充图4中,特征基因网络的层次聚类树状图表示模块之间以及临床特征权重之间的关系。
鉴定和验证关键基因
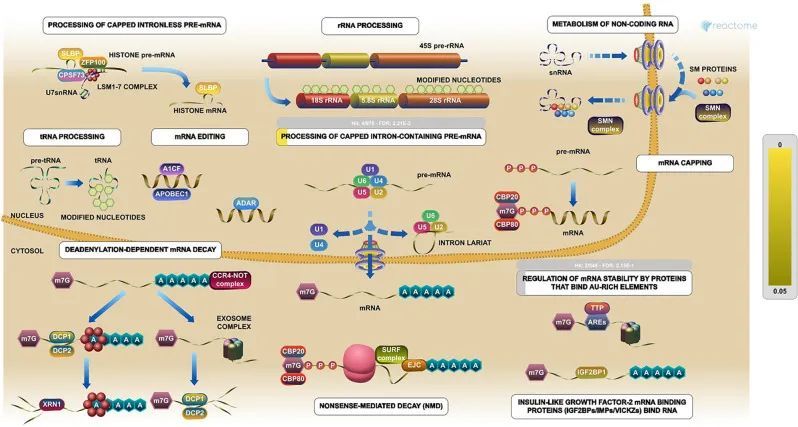
使用STRING数据库构建了由403个差异表达基因(DEGs)组成的蛋白质相互作用网络(PPI网络)。通过Cytoscape插件(MCODE、Cytoscape和CytoHubba)确定了整个网络中关键基因之间的交互关系。根据得分系统(截断k-score = 12),从MCODE中鉴定出两个亚型:亚型1有82个节点和938条边,亚型2有20个节点和168条边。此外,数据还被导入到另一个插件CytoHubba中,通过EPC、MCC、DMNC、MNC和Stress等五种不同的计算方法,帮助鉴定出104个关键基因。然后,这两个亚型被导入到CytoNCA插件中,通过betweeness、closeness、degree、eigenvector和subgraph等五种不同的算法,帮助鉴定出40个关键基因。作者坚信,这些关键基因是PPI网络和WGCNA分析中与表型(性别)高度相关的207个基因(附表1)之间的交集(图4A、B)。最后,在BE和EAC之间确定了五个SHGS,即,pre-mRNA加工因子4(PRPF4),丝氨酸和精氨酸富集剪接因子1(SRSF1),异核核糖核蛋白M(HNRNPM),DExH盒螺旋酶9(DHX9)和起源识别复合物亚单位2(ORC2)。通路富集分析表明,所有的SHGS都参与RNA代谢,其分子功能术语包括细胞周期、DNA结合、DNA拓扑异构酶结合、pre-mRNA剪接和RNA螺旋酶活性(图5)。

生存分析和免疫组织化学
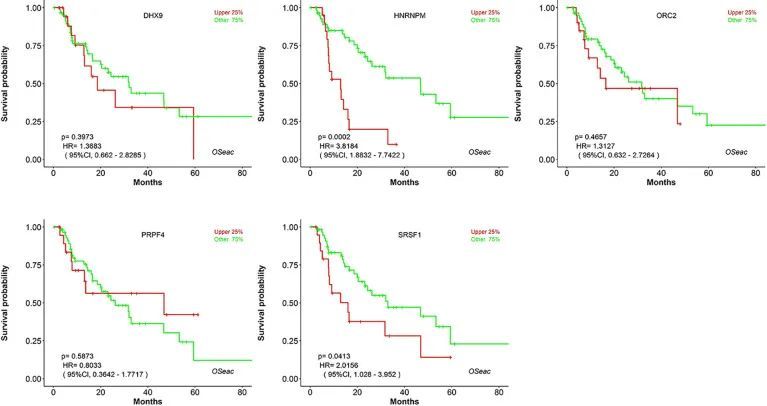
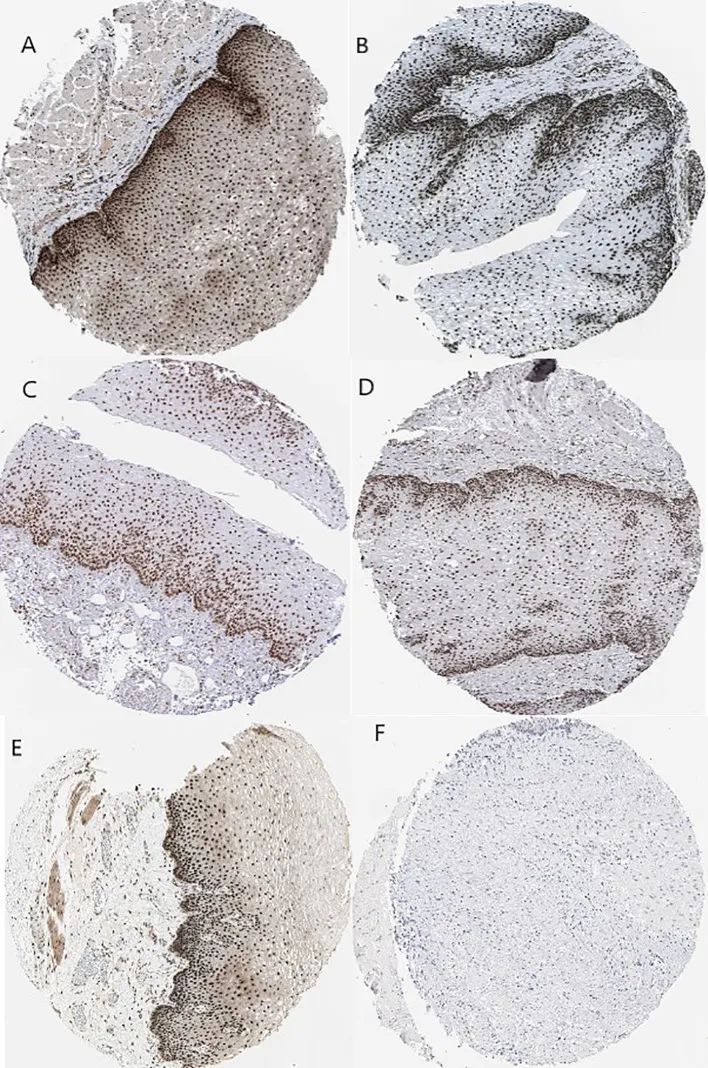
Kaplan-Meier图表显示了SHGS的预后影响,这些影响来自PPI网络复合物和WGCNA的模块。结果显示,HNRNPM和SRSF1的高表达与BE和EAC患者的整体生存率较差相关(p < 0.05)。此外,PRPF4、DHX9和ORC2的高表达与BE和EAC患者的整体生存率较长相关(图6)。此外,作者绘制了一个基于性别的生存曲线来确定WGCNA模块的相关性。在男性中,危险比(HR)和95%置信区间如下:PRPF4(HR = 1.08;95%CI - 0.46 ± 2.48;p = 0.865);SRSF1(HR = 3.08;95%CI - 1.49 ± 6.37;p = 0.002);HNRNPM(HR = 3.295;95%CI - 1.54 ± 7.02;p = 0.002);DHX9(HR = 1.39;95%CI - 0.64 ± 2.48;p = 0.404);ORC2(HR = 1.25;95%CI - 0.58 ± 2.72;p = 0.564)。此外,在女性病例中,PRPF4(HR = 0.39;95%CI - 0.03 ± 3.89;p = 0.421);SRSF1(HR = 1.49;95%CI - 0.20 ± 10.79;p = 0.689);HNRNPM(HR = 8.06;95%CI - 0.82 ± 79.01;p = 0.073);DHX9(HR = 0.38;95%CI - 0.04 ± 3.89;p = 0.424);ORC2(HR = 3.24;95%CI - 0.20 ± 51.91;p = 0.4061)。结果清楚地表明,与女性相比,SHGS的高表达与男性的预后不良相关。此外,人类蛋白质图谱数据库的免疫组化切片显示,与相邻正常组织相比,癌组织中SHGS的蛋白表达显著增加,如图7所示。因此,这些SHGS都是起着主动作用并可能具有共同表达倾向的关键基因。
总结
这项基于网络药理学的研究为BE和EAC患者的诊断和预后提供了新的见解。基于微阵列数据集的PPI网络和WGCNA的结果表明,深灰色模块与EAC和BE的相关性最大,识别出五种SHGS,即PRPF4、SRSF1、HNRNPM、DHX9和ORC2。基于WGCNA的基因共表达网络表明,共表达基因与临床特征(患者性别)之间的关系与癌症的进展有关。