文章目录
- RNA折叠
- RNA folding representation
- 1 DP for simple folds
- 1.1 Nussinov Algorithm objective
- 1.2 energy constraints
- 1.3 The key idea of the algorithm
- 2 DP for stacking and complex folds
- Stochastic context free grammars
来自Manolis Kellis教授(MIT计算生物学主任)的课
油管链接:6.047/6.878 Lecture 7 - RNA folding, RNA world, RNA structures (Fall 2020)
本节课分为三个部分,本篇笔记是第三部分RNA folding。
本课程探讨RNA的折叠过程及其预测方法。先了解了RNA的表示和Nussinov算法的应用。然后通过动态规划预测复杂的RNA叠加与折叠。最后介绍了随机上下文无关文法,为RNA结构提供了广泛的描述框架。
RNA折叠

RNA折叠是一个过程,其中RNA分子将自己折叠成特定的三维结构,这些结构对于其功能至关重要。
RNA folding representation
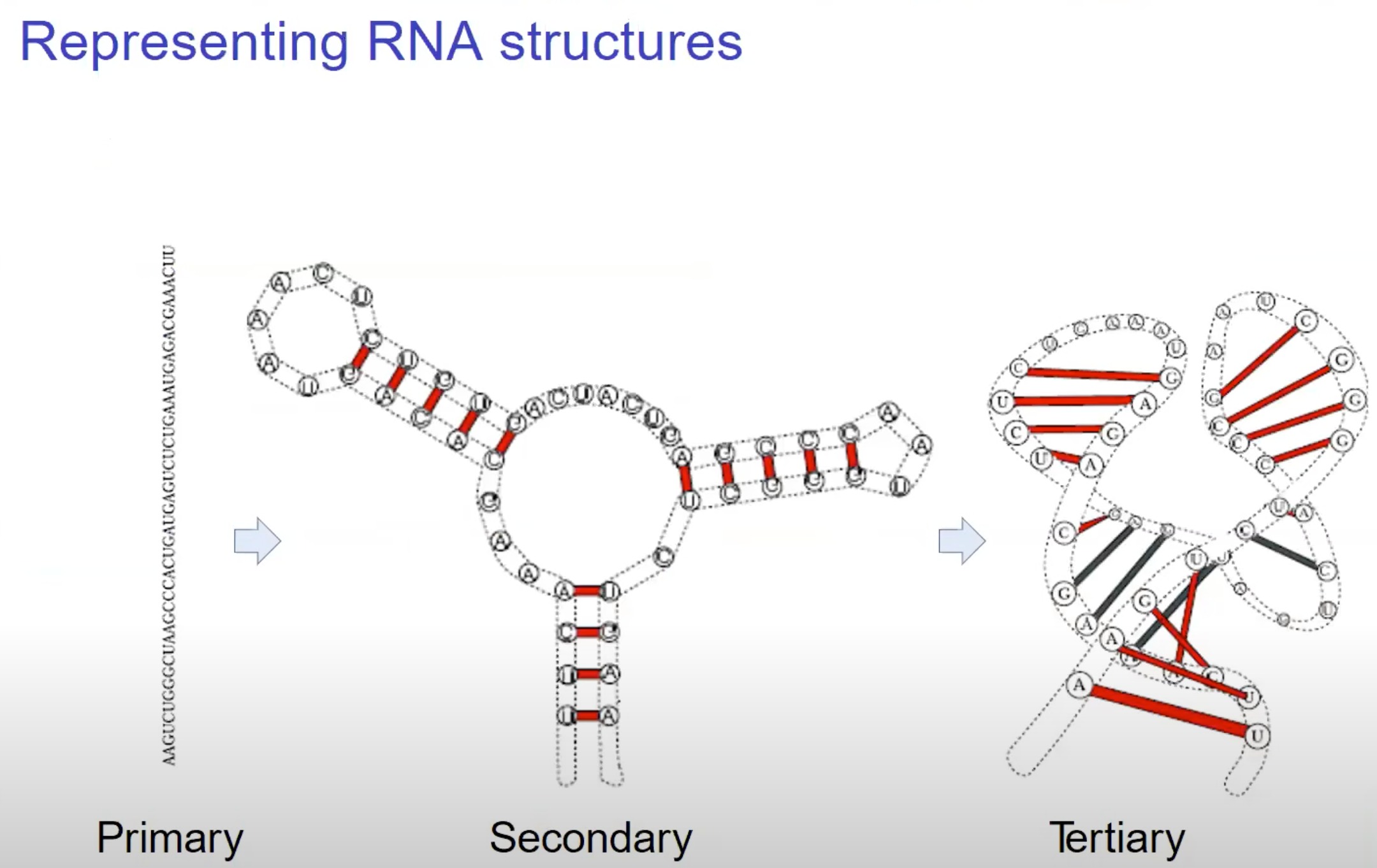
- 初级结构 (Primary)
- 二级结构 (Secondary): 二级结构展示了RNA分子如何在空间中折叠并通过氢键形成基对。tRNA和rRNA就是依赖其二级结构来发挥作用的。
- 某些RNA的功能区域可能是由其特定的二级结构模式决定的。
- 三级结构 (Tertiary): 三级结构描述了RNA分子如何进一步在三维空间中折叠,以形成更复杂的结构。在这一级,分子的不同部分可能会通过疏水相互作用、范德华力和其他非共价相互作用来相互接触。

- 能量的覆盖: RNA二级结构占据了折叠过程中的大部分自由能。这意味着RNA的稳定性和形成方式在很大程度上取决于其二级结构。
- 生化粗粒化: RNA的二级结构为我们提供了一个粗略的、生化上有意义的3D结构表示。这使得研究人员可以更容易地理解RNA的空间排布和功能性质。
- 折叠的中间态: 在形成其完整的三维结构之前,RNA首先会形成一个稳定的二级结构。这是RNA折叠成其最终功能形态的关键步骤。
- 进化上的保守性: 在进化过程中,RNA的二级结构往往是保守的。这意味着相似的二级结构在不同的生物中可能具有相似的功能。
- 易于计算处理: 与RNA的完整三维结构相比,其二级结构相对简单,因此更容易在计算机上建模和分析。
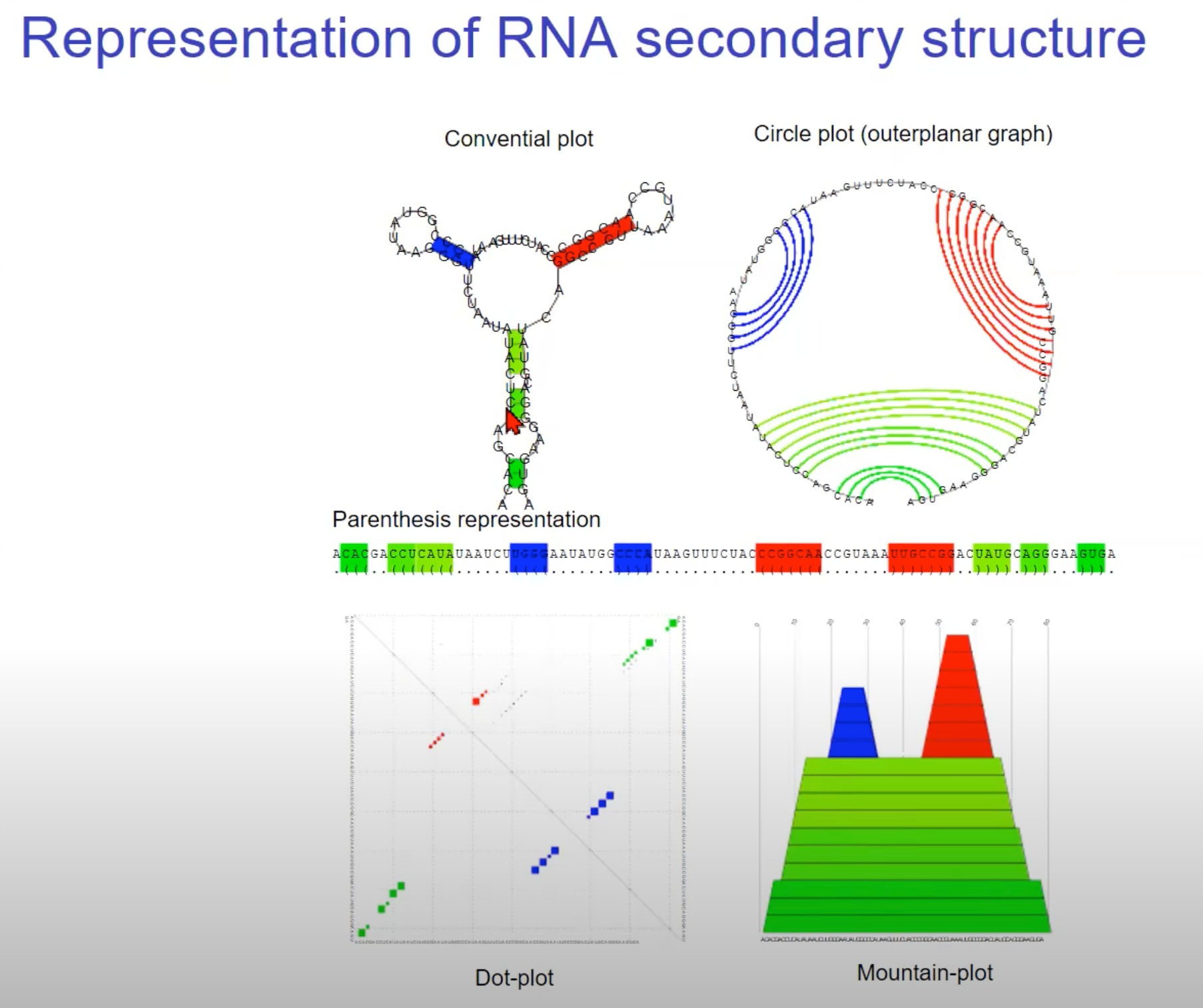
这个图展示了RNA二级结构的不同表示方法。以下是每种表示方法的简要解释:
-
常规图(Conventional plot): 这种图形展示了RNA分子的实际形状和其碱基配对。
-
圆形图(Circle plot/outerplanar graph): 这是RNA二级结构的另一种表示方法,其中RNA序列被放置在一个圆上,碱基配对则由连接线表示。这种方法适用于显示复杂的或交叉的碱基配对关系。
-
括号表示法(Parenthesis representation): 在这种表示法中,RNA序列从左到右写出,而碱基配对则通过括号表示。匹配的左括号和右括号表示一对碱基配对,而点表示未配对的碱基。这种表示法非常紧凑,适用于计算和算法处理。
-
点图(Dot-plot): 点图显示了RNA序列内部所有可能的碱基配对。图上的每个点代表一个特定的碱基配对。这种方法常用于预测RNA二级结构和比较不同结构的相似性。
-
山形图(Mountain-plot): 这种表示法通过高度变化来描绘RNA的二级结构。高度的变化反映了RNA结构中的堆叠区域。这提供了一个直观的方式来了解RNA的叠层结构和拓扑形态。
1 DP for simple folds
使用动态规划(Dynamic Programming)来解决RNA折叠问题。RNA折叠是预测RNA分子在三维空间中如何折叠成其稳定的结构的问题。
- 当RNA分子折叠时,某些碱基对之间会形成氢键,形成了所谓的碱基配对。我们通常希望优化的是整个结构的稳定性,这可以通过计算配对得分来评估
- 参考我们前面序列对比的动态规划,尝试应用在这里
1.1 Nussinov Algorithm objective

算法目标:预测RNA的二级结构
-
Objective function(目标函数):
- Nussinov算法的目标是找到具有最大嵌套配对数的RNA结构。所谓的嵌套配对意味着这些配对不与其他配对交叉。
-
Recursive computation(递归计算):
- Use dynamic programming?
- How?:具体实施方式涉及建立一个得分矩阵,并通过考虑RNA序列中每个可能的碱基对来填充这个矩阵。每个位置的得分都是基于它可以形成的最佳配对,以及它与其他碱基之间的关系。
-
Similarities to sequence alignment(与序列比对的相似性):
- 在序列比对中,我们试图找到两个或多个序列之间的最佳对齐方式。对于RNA结构预测,我们试图找到RNA序列内部的最佳对齐方式,即碱基配对方式。
1.2 energy constraints

RNA folding based on biophysical energy constraints(基于生物物理能量约束的RNA折叠)
-
Scoring scheme(评分方案):
- 为了决定RNA的最优结构,这里有一个评分系统。在物理上,结构的优化通常是基于最小自由能的。自由能在这里代表稳定性,较低的自由能意味着更稳定的结构。
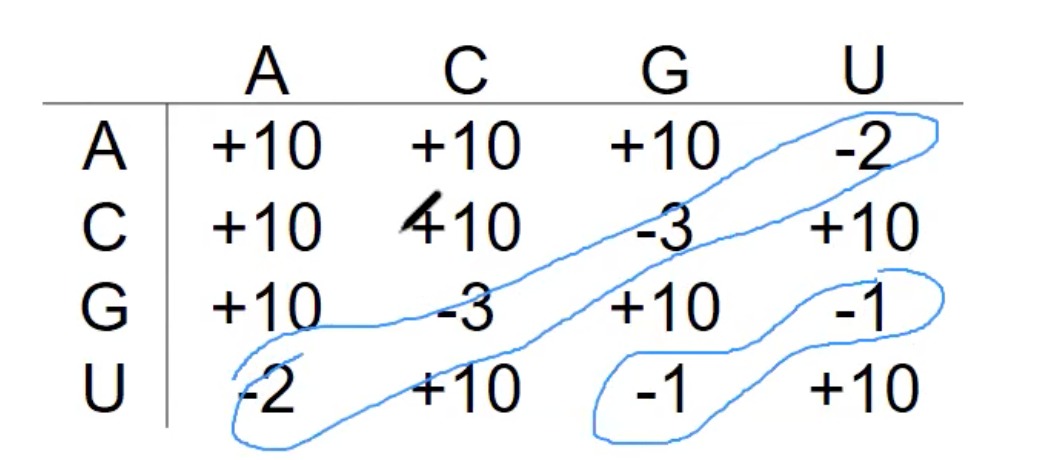
- "-1"表示G和U的配对,特殊
- 为了简化模型,为每对碱基配对分配了一个能量值。例如,A与U的配对有-2的能量值,而C与G的配对有-3的能量值。这些值可能是基于实验数据的。
-
Does DP apply?(动态规划是否适用?):
- 考虑到能量分数是可加的。
- 稍后可能会讨论更复杂的能量模型,但基本的思路是,通过排除某些结构(如伪节点),可以将问题分解为子问题,这使得动态规划变得可行。
-
Define “matrix”: Eij:
- 这定义了一个矩阵E,其中Eij表示子序列i到j之间的最小能量。
- 这个矩阵用于动态规划算法,储存每个子序列的最优解,从而避免重复计算。
-
Recursion formula(递归公式) 和 Traversal order(遍历顺序)
通过为每对碱基配对分配能量分数,并使用动态规划来找到具有最低总自由能的结构,我们可以获得RNA的预测结构。
1.3 The key idea of the algorithm

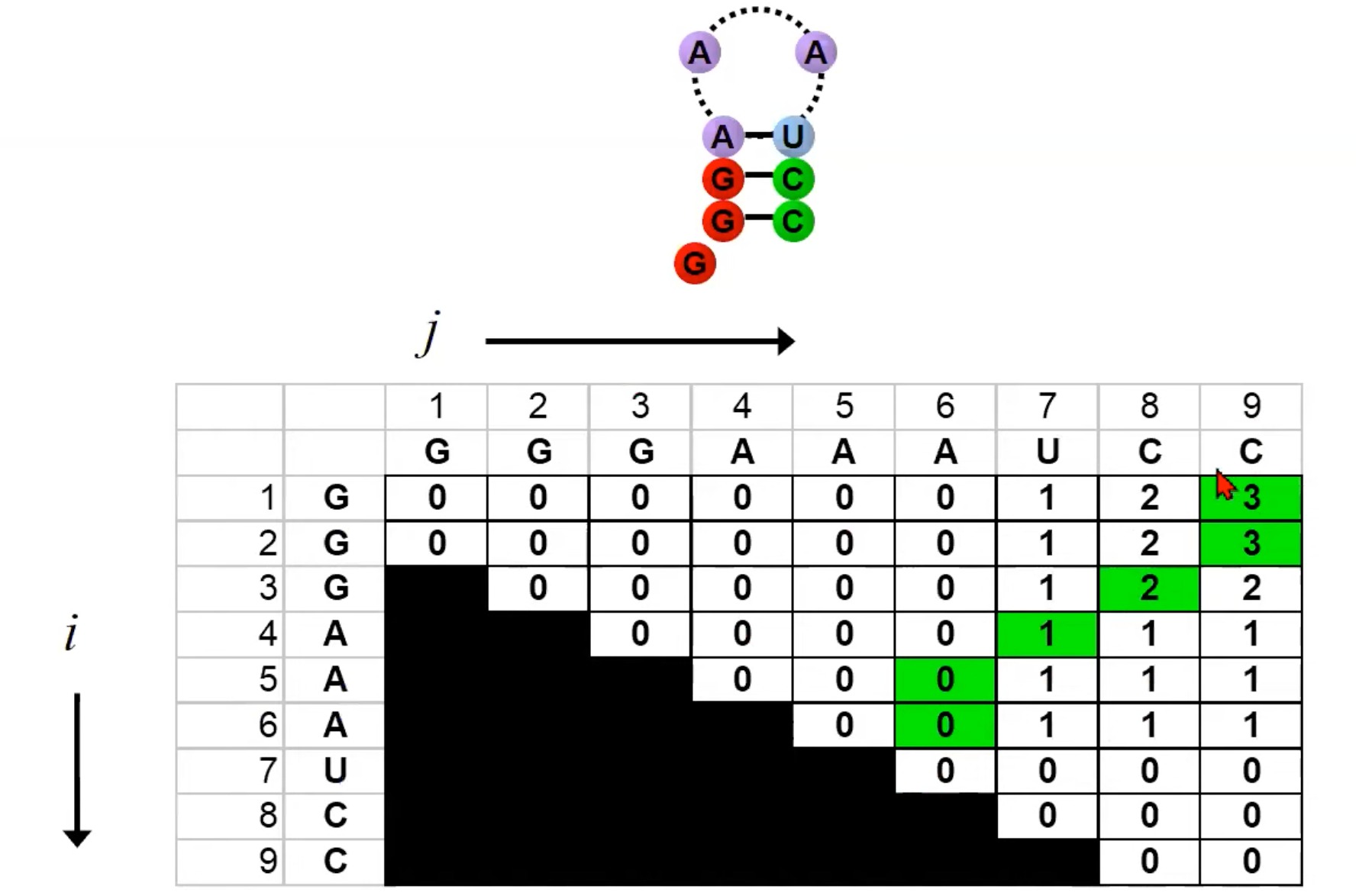
右侧的图显示了一个填充了数字的大矩阵。这个矩阵的填充表示算法的动态规划过程。每个矩阵的单元格代表一个RNA子序列的得分。为了找到整个RNA序列的最优二级结构,算法会逐步填充这个矩阵。
-
矩阵的对角线:矩阵的对角线上的值都是0,因为一个单独的碱基不能与自己形成配对,所以得分为0。
-
填充顺序:可以看到图中的蓝色箭头,这表示填充矩阵的顺序。算法从对角线开始,然后向右上方移动,填充每一个子矩阵。
-
碱基配对:在图中,有些地方用线连接了两个点。这表示在那个特定的RNA子序列中,两个碱基形成了配对。
-
计算得分:当计算
F(i, j)时,我们会查看以下两种情况:i和j配对的得分(如果它们可以配对的话)加上子序列F(i+1, j-1)的得分。- 不考虑
i和j的配对,而是查看从i到j之间所有可能的分割点k,然后取F(i, k)和F(k+1, j)的得分之和的最大值。 - 下一个遍历是往右上角遍历
-
该矩阵的填充反映了RNA序列的所有可能的二级结构。算法通过比较所有这些可能性来找到得分最高的结构。

i , j i,j i,j配对:
- 在这种情况下,位置 i i i的碱基与位置 j j j的碱基形成了配对,因此它们之间形成了一个碱基对。
- 同时,子序列 x i + 1 x_{i+1} xi+1 到 x j − 1 x_{j-1} xj−1 可能还有其他的最佳配对,记为 F ( i + 1 , j − 1 ) F(i+1, j-1) F(i+1,j−1)。
i i i未配对:
- 在这里,位置 i i i的碱基没有与任何其他碱基配对。
- 子序列 x i + 1 x_{i+1} xi+1 到 x j x_j xj 继续寻找其他可能的最佳配对。
j j j未配对:
- 与“ i i i未配对”的情况类似,这里位置 j j j的碱基没有与任何其他碱基配对。
- 子序列 x i x_i xi 到 x j − 1 x_{j-1} xj−1 继续寻找其他可能的最佳配对。
分叉:
- 在这种情况中,序列在某个位置 k k k处被分成了两部分。
- 从 x i x_i xi 到 x k x_k xk 的子序列和从 x k + 1 x_{k+1} xk+1 到 x j x_j xj 的子序列分别寻找自己的最佳配对。
- 这是一个更复杂的情况,因为我们需要考虑从 i i i到 j j j之间的所有可能的 k k k值,并为每个 k k k值计算其得分 F ( i , k ) + F ( k + 1 , j ) F(i, k) + F(k+1, j) F(i,k)+F(k+1,j)。最后,我们选择使得 F ( i , j ) F(i, j) F(i,j) 得分最大的 k k k值。

2 DP for stacking and complex folds
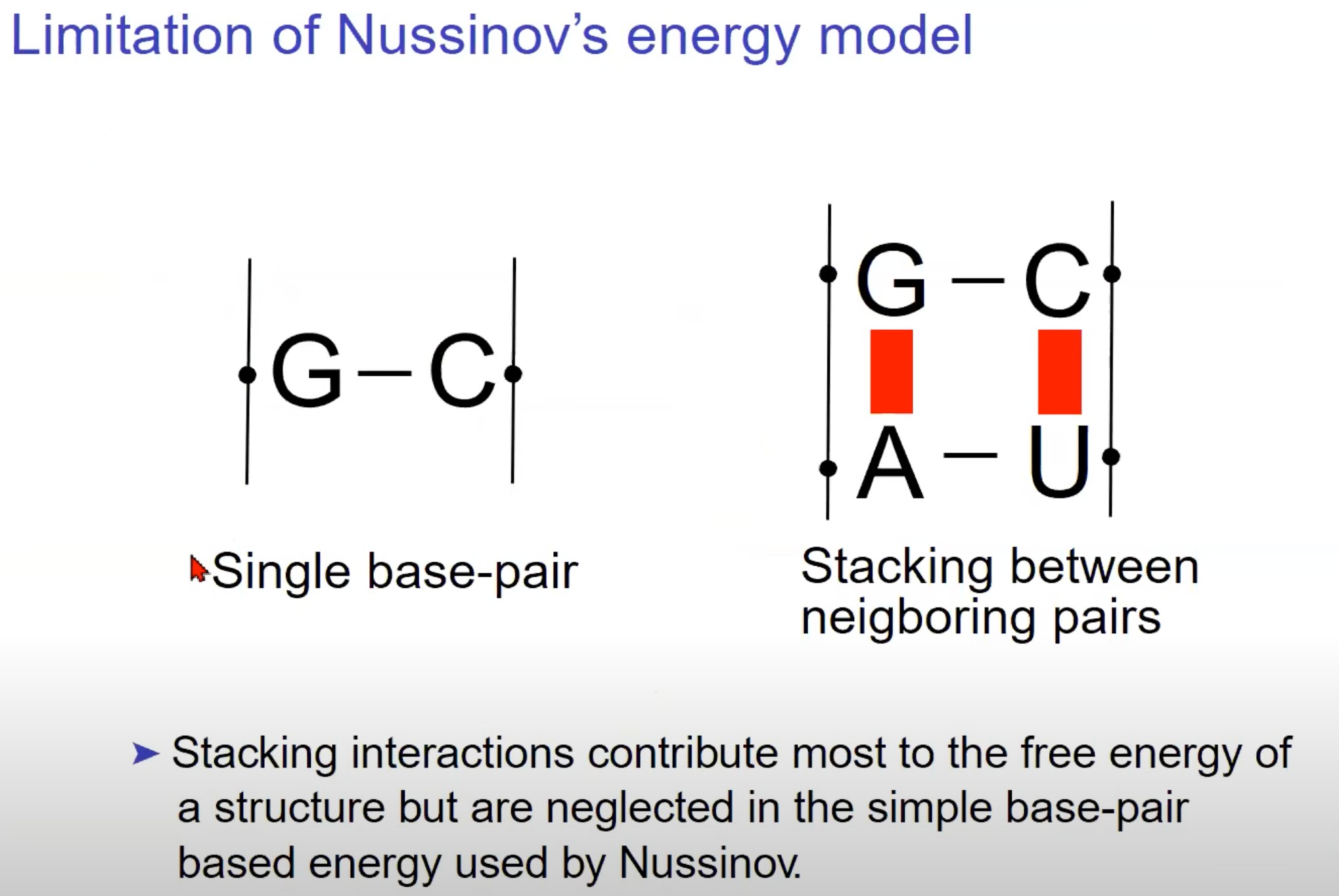
Nussinov能量模型的局限性
- 除了单个碱基对外,RNA碱基还倾向于在空间中堆叠,形成堆叠结构。这里显示了一个G-C配对和一个A-U配对的堆叠。堆叠交互是由于相邻碱基对之间的香族环的π-π交互。
- 堆叠交互为RNA分子提供了额外的稳定性,因此它们对RNA分子的整体自由能有着重要的贡献。
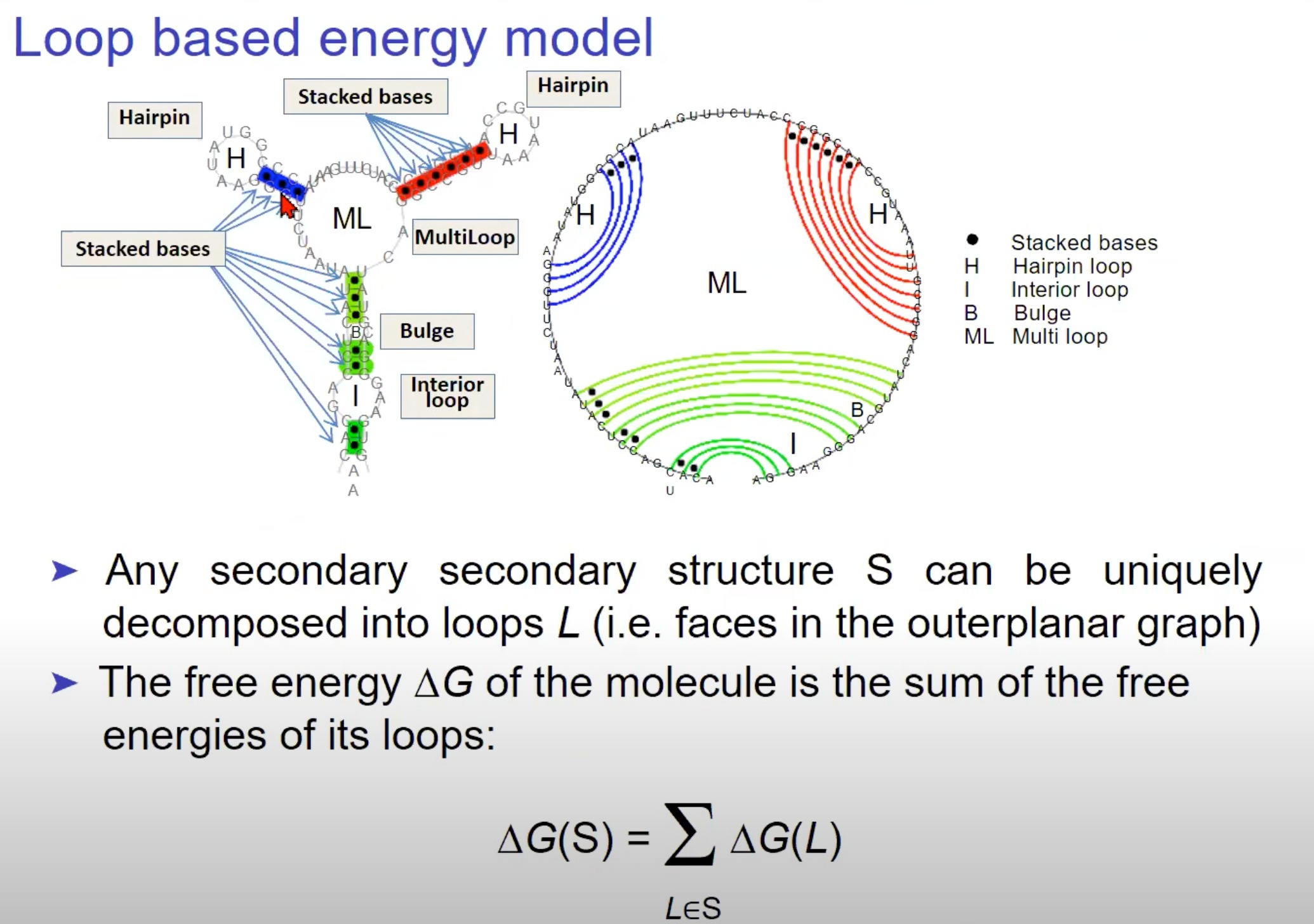
- 基于环(Loop)的能量模型
- 堆叠的碱基(Stacked bases)、发夹环(Hairpin loop)、内部环(Interior loop)、Bulge、以及多环(Multi loop)。这些是RNA二级结构的基本组成部分。
“任何二级结构 ( S ) 都可以被唯一地分解成多个环 ( L )”:这意味着任何RNA的二级结构都可以被唯一地分解为环。
- “分子的自由能 ( $\Delta G $) 是其环的自由能之和”:整个RNA分子的自由能是由它的各个环的自由能组成的。
- 这可以用以下公式表示:
$
\Delta G(S) = \sum_{L \in S} \Delta G(L)
$
这表示分子 ( S ) 的自由能是它包含的所有环 ( L ) 的自由能的总和。
扩展补充:
- Structural ensemble and base-pairing probability
二维的点图,用于可视化RNA分子中可能的碱基配对的概率


在室温下,RNA不仅形成一个结构,而是存在为一系列可能的结构,称为集合(ensemble)
使用热力学理论来计算特定结构的概率
Stochastic context free grammars



上下文无关文法 (CFG),用于生成回文:
V
=
{
S
}
,
T
=
{
a
,
b
}
,
P
=
{
S
→
a
S
a
,
S
→
b
S
b
,
S
→
ϵ
}
V = \{S\}, \quad T = \{a, b\}, \quad P = \{S \rightarrow aSa, S \rightarrow bSb, S \rightarrow \epsilon\}
V={S},T={a,b},P={S→aSa,S→bSb,S→ϵ}
这个CFG可以有效地描述字母表 ({a, b}) 上的所有回文。
RNA文法的例子。这是一个上下文无关文法,旨在模拟RNA的二级结构。RNA分子由四种碱基组成:A(腺苷)、C(鸟苷)、G(鸟嘌呤)和U(尿嘧啶)。
V
=
{
S
}
,
T
=
{
a
,
c
,
g
,
u
}
,
P
=
{
S
→
a
S
u
,
S
→
c
S
g
,
…
}
V = \{S\}, \quad T = \{a, c, g, u\}, \quad P = \{S \rightarrow aSu, S \rightarrow cSg, \dots\}
V={S},T={a,c,g,u},P={S→aSu,S→cSg,…}
这里的产生规则模拟了RNA的碱基配对,例如A与U配对,C与G配对。
解析树 (Parse Tree) 描述了如何使用给定的文法来派生特定的RNA序列。在这个例子中,给定的RNA序列是 (ACAGGAACUGUACGCUGCACCGC),解析树展示了如何从初始非终结符 (S) 开始,通过多次应用产生规则来得到这个序列。
此外,RNA的二级结构图展示了该序列的配对模式,其中一些碱基通过氢键配对形成稳定的结构。




![[Docker]一.Docker 简介与安装](https://img-blog.csdnimg.cn/b240c91596204ffa974936dc021e0129.png)