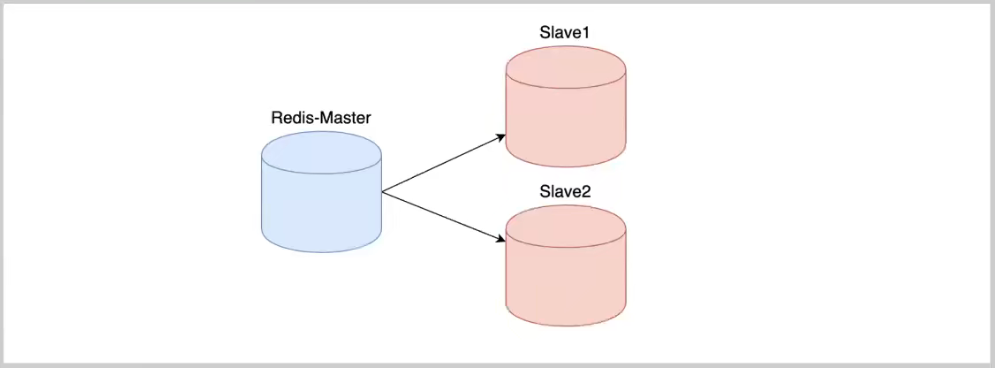
多肽是两个以上氨基酸通过肽键组成的生物活性物质,可以通过折叠、螺旋形成更高级的蛋白质结构。多肽不仅与多个生理活动相关联,还可以自组装成纳米粒子,参与到生物检测、药物递送、组织工程中。
然而,多肽的序列组成过于多样,仅 10 个氨基酸就可以组成超过百亿种多肽。因此,人们很难对其自组装特性进行全面系统的研究,进而优化自组装多肽的设计。
为此,西湖大学的李文彬课题组利用基于 Transformer 的回归网络,对百亿种多肽的自组装特性进行了预测,并分析得到了不同位置氨基酸对自组装特性的影响,为自组装多肽的研究提供了强力的新工具。
作者 | 雪菜
编辑 | 三羊
多肽是两个以上氨基酸通过肽键组成的生物活性物质。多肽合成便利、可生物降解、生物相容性强,且具有丰富的化学多样性,可以组成具有荧光、半导体导电性或是磁性的纳米物质。正因为此,多肽得到了科研界的广泛关注。
然而,也正是因为多肽的多样性,人们暂缺乏预测其自组装倾向 (AP, Aggregation Propensity) 的方法,很难将其转变为有序结构。目前只有极少的多肽能够自组装,形成满足需求的超分子结构,并投入到工业应用中。

过去数十年间,自组装多肽主要是通过生物实验发现的。然而,实验往往需要很长的周期,而且存在一定的倾向性,不利于对大量多肽进行全面的系统研究。
近年来,计算筛选 (Computational Screening) 被广泛用于自组装多肽的设计中。2015 年,Frederix 等人利用粗粒度分子动力学 (CGMD) 分析了三肽的 AP。然而,随着氨基酸数量的增加,多肽序列数量会指数级增长,使 CGMD 的成本大幅增加。
因此,有研究者结合 AI 和 CGMD ,以降低传统方法的分析成本。然而,AI-CGMD 需要大量的训练数据。据推测,十肽 (decapeptide) 的序列超过百亿种,需要 320 万个多肽序列数据。基于上述原因,目前尚无对 5 个以上氨基酸组成多肽 (pentapeptide) 的 AP 预测。
为解决这些问题,西湖大学的李文彬课题组利用基于 Transformer 的回归网络 (TRN),结合 CGMD,对百亿种多肽的自组装特性进行了预测,得到了五肽和十肽的 AP,并得到了不同位置的氨基酸对多肽 AP 的影响。这一成果已发表于「Advanced Science」。

论文链接:
https://onlinelibrary.wiley.com/doi/full/10.1002/advs.202301544
实验过程
训练集:拉丁超立方采样
首先,利用拉丁超立方采样筛选出 8,000 个多肽序列。筛选出的多肽序列通过 CGMD 模型分析得到其 AP。
模型构建:编码与解码
研究人员基于 TRN 构建了 AP 预测模型。模型包括 Transformer 编码器和多层感知机 (MLP) 解码器。Transformer 编码器由输入嵌入层 (Input Embedding)、位置编码器 (Positional Encoding) 和编码块 (Encording Block) 组成。
输入嵌入层用于将多肽的组成单元(即氨基酸)映射到 512 维的连续空间中,位置编码器会输出氨基酸的位置信息。编码块包括自注意网络和前馈神经网络。
Transformer 编码器最终输出一个隐藏层表示的多肽序列。这一序列经过 MLP 降维 5 次后,被压缩为一维向量。MLP 解码器的最后一层会输出多肽的 AP。

a:α-螺旋和 β-折叠的原子模型及 α-螺旋的 CG 模型;
b:通过 CGMD 输出训练数据的流程;
c:TRN 模型示意图。
实验结果
模型预测:提升 54.5%
研究人员对比了 TRN 模型和其他非深度学习模型(支持向量机 SVM、随机森林 RF、临近算法 NN、贝叶斯回归 BR 和线性回归 LR)的 AP 预测表现。
在仅有 8,000 个训练数据时,模型的决定系数 R2 就超过了 0.85,较 SVM 提升了 11.8%,较 RF 提升了 54.5%

随着训练数据的增加,TRN 模型的表现随着增加。 当训练数据达到 54,000 时,TRN 模型的平均绝对误差 (MAE) 为 0.05,R2 为 0.92。

上述结果说明,相比非深度学习模型,TRN 模型可以用较少的训练数据达到较高的预测率。同时,随着训练数据的增加,TRN 模型的表现随之提升。
亲水性:APHC 修正
据报道,除 AP 外,多肽的亲水性 (log P) 也会对多肽的自组装产生影响。
当 AP 自低向高增长时,log P 的中位数随之降低,说明亲水性强的多肽聚集能力较差。然而,log P 位于 0.25-0.75 之间的多肽 AP 跨度很大,分布在 0-1 之间,说明二者的联系并不密切,还有其他因素会影响多肽的 AP。

a:320 万种五肽的 AP 与 log P 的相关性;
b:AP 在不同区间的分布;
c:log P 在不同 AP 区间的分布。
为找出 AP 和 log P 对多肽自组装的影响,研究人员利用 log P 对 AP 进行了修正,得到了 APHC。修正后的 APHC 能够分辨出多肽自组装和沉淀,筛选出可以形成水凝胶的多肽。

图 6:APHC 与 log P 的关系
a:320 万种五肽的 APHC 与 log P 的相关性;
b:APHC 在不同区间的分布;
c:log P 在不同 APHC 区间的分布。
自组装法则:不同位置的氨基酸影响
在分析了五肽中不同位置的 20 种氨基酸对 APHC 的影响后,研究人员总结得到了不同氨基酸及其分布对多肽自组装特性的影响,并将其分成了 5 组。
第一组氨基酸包括苯丙氨酸 (F)、酪氨酸 (Y) 和色氨酸 (W)。这组氨基酸中存在 π-π 堆叠且疏水性强,对多肽自组装贡献最大。其中 W 的疏水性最强,对 APHC 的影响最大,这与 WWWWW 的观察结果一致。

F、Y、W 在 3-5 号位,尤其是 3 号位时,对多肽自组装贡献最强。可能是因为在 3 号位上,氨基酸的自由度较高,更易通过 π-π 作用驱动多肽自组装。

然而,这些芳香类氨基酸在 5 号位时,是强质子接受体,会与其他多肽相互作用,拉大苯环的距离,削弱分子内的 π-π 作用。
第二组氨基酸包括异亮氨酸 (I)、亮氨酸 (L)、缬氨酸 (V) 和半胱氨酸 © 。由于这些氨基酸的侧链和水之间相互排除,疏水性强,对多肽自组装贡献较强。这组氨基酸常分布在多肽的两端,尤其是自组装多肽的 N 端。

第三组氨基酸包括组氨酸 (H)、丝氨酸 (S) 和苏氨酸 (T)。这组氨基酸有极化侧链,可以通过氢键提升多肽的自组装能力。然而,氢键的作用相比于 π-π 堆叠较弱,因此在高 APHC 的多肽中,第三组氨基酸含量较少。
T 和 S 倾向于占据多肽的两端,尤其是 N 端,这有利于氢键的形成。而 H 会远离多肽的两端。

第四组氨基酸包括蛋氨酸 (M) 和脯氨酸 § 。M 和 P 在不同 APHC 的多肽中分布基本一致,仅对多肽的特定指标有微弱的影响。
第五组氨基酸不利于多肽的自组装,包括带负电的天冬氨酸 (D) 和谷氨酸 (E)、带正电的赖氨酸 (K) 和精氨酸 ®、强极性的天冬酰胺 (N) 和谷氨酰胺 (Q)、无侧链的丙氨酸 (A) 和甘氨酸 (G)。
然而,C 端的 D 和 E、N 端的 R 和 K 可以形成带双电荷的头基,通过异性电荷相互吸引、形成盐桥促进多肽的自组装。N 和 Q 由于极性太强,会促进多肽的溶解。而 A 和 G 缺乏明显的相互作用,不利于多肽自组装。

实验验证:与 CGMD 和 TEM 结果基本一致
为确认 TRN 模型的预测结果,研究人员用 CGMD 对五种多肽的自组装特性进行了验证。CGMD 的计算结果与 TRN 模型的预测结果基本一致。
同时,NRMMR、DMGID、NRMMRDMGID 和 NRMMR + DMGID 的自组装特性还得到了实验的验证。透射电子显微镜 (TEM) 的结果与 CGMD 的结果基本一致。

上述结果说明,TRN 模型可以准确预测五肽、十肽和混合五肽的自组装特性,为自组装多肽的研究提供了强力的新工具。
自组装多肽:生物医药新方向
虽然人们对多肽的自组装特性研究还不够深入,但自组装多肽已经广泛用于组织工程、药物递送和生物传感当中。此外,细胞的收缩和舒张、内吞囊泡的移动、细菌和病毒的跨膜传输都离不开多肽的自组装,阿尔兹海默症、帕金森氏病和II型糖尿病等疾病也与蛋白质的错误折叠有关。

随着 AI 的发展,科研人员对于大批量数据的处理能力不断增强。生物研究从传统的实验研究,走向计算研究,再走向 AI 研究的同时,研究的规模也从以往的几十上百种可能,逐渐迈向了百亿种。在 AI 的帮助下,人类正在推进生物研究的边界,相信未来人们能对生物有更精细更全面的研究,让 AI + 生物普惠大众。
参考链接:
https://pubs.rsc.org/en/content/articlelanding/2014/CS/C4CS00161C