非酒精性脂肪性肝炎(NASH)是非酒精性脂肪性肝病(NAFLD)的晚期亚型,已成为肝硬化和肝细胞癌等终末期肝病最重要的病因。本研究旨在探索与非酒精性脂肪肝相关的新基因。
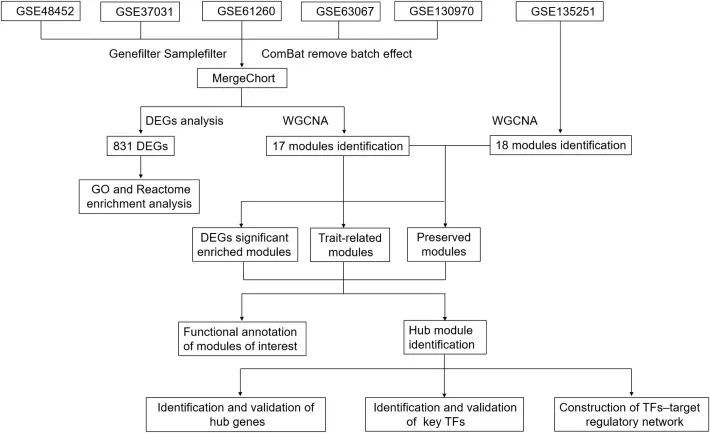
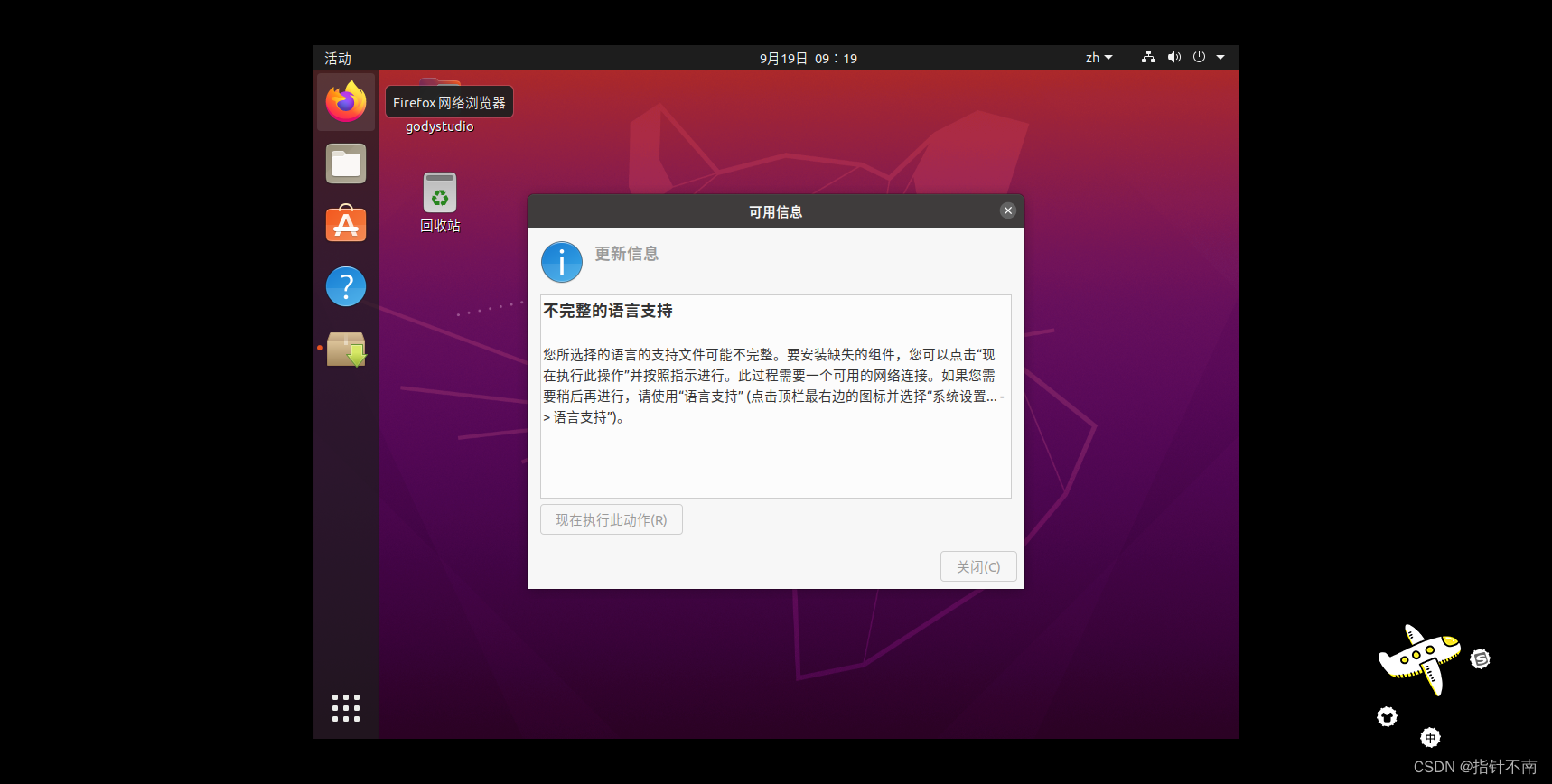
图1 流程图
1.收录的 GEO 数据集信息
根据之前制定的纳入标准,本研究纳入了 GSE48452、GSE37031、GSE61260、GSE63067 和 GSE130970。这五个数据集中有 104 例 NASH 患者和 70 例对照。在去除离群值后,97 名 NASH 患者和 67 名对照组被保留在下面的分析中。为了消除来自不同平台和批次的水浴效应,作者使用战斗函数消除了五个数据集的批次效应。通过合并不同平台,共检测到 12579 个基因。在消除批次效应之前,根据归一化前表达值的前两个主成分(PCs)对样本进行批次分组。而将五个平台的样本合并后,根据主成分分析,样本的整体表达量呈均匀分布,表明不同平台造成的影响分子生物学差异估计的批次效应已被成功纠正。此外,本研究还使用数据集 GSE135251 作为验证数据集。
2.NASH 患者 DEGs 的鉴定
五组合并数据集(MergeCohort)的基因表达矩阵的主成分分析图见图 2A。在合并队列中,对照组和 NASH 组共鉴定出 831 个 DEGs(本杰明-霍赫伯格调整 p 值< 0.05, absolute difference of mean > 0.3),其中包括 600 个上调和 231 个下调 DEGs(图 2B)。
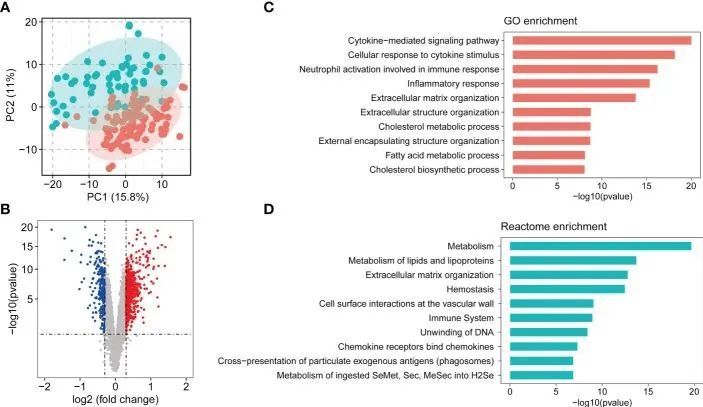
图2 健康对照组和非酒精性脂肪性肝炎(NASH)患者的基因表达谱组合概况
3.DEG 的功能和通路富集分析
在本研究中,作者进行了GO和Reactome通路富集分析,以确定831个DEGs在NASH发病机制中的潜在功能。生物过程分析(图 2C)显示,在 NASH 中,这些基因与多个免疫相关通路有关,如细胞因子介导的信号通路、细胞对细胞因子刺激的反应以及参与免疫反应的中性粒细胞活化。细胞外基质组织和细胞外基质结构组织等一些与 ECM 相关的通路也被富集。此外,还发现了胆固醇代谢过程、脂肪酸代谢过程、胆固醇生物合成过程和其他生物过程等代谢过程。通过 Reactome 通路分析,研究了基于 DEGs 的通路。前 10 个通路如图 2D 所示。其中,代谢、脂类和脂蛋白的代谢、细胞外基质组织、免疫系统、趋化因子受体结合趋化因子被显著富集。因此,上述结果表明,代谢、细胞外基质相关通路和免疫相关通路在 NASH 的发生和发展过程中起着重要作用。
4.WGCNA 和确定与 NASH 疾病状态相关的模块
为了捕捉与 NASH 状态相关的离散共表达基因群,并将已确定的表达差异整合到更高的系统水平背景中,作者根据合并队列中前 5000 个中位绝对偏差(MAD)基因进行了共表达网络分析(WGCNA)。根据无标度拓扑标准,本研究中考虑了β=5(图 3A)。根据动态树切割,分层聚类树枝图产生了 17 个不同的基因模块,如图 3B 所示。有 909 个基因未能归入一个不同的组,被归入灰色模块,本研究忽略了这一模块。模块的大小从 86 个(Grey60 模块)到 734 个(Turquoise 模块)不等(图 3C)。图 3D 显示了各模块中 DEGs 的富集情况,其中上调基因主要显著富集在绿松石模块(n = 233,p = 1.93 × 10 -44 ),其次是青色模块(n = 54,p = 1.24 × 10 -15 )、灰色 60 模块(n = 40,p = 2.05 × 10 -13 )、茶色(n = 48,p = 1.59× 10 -9 )和品红色(n = 47,p = 2.77× 10 -4 ),下调基因在黑色(n = 107,p = 9.25× 10 -86 )和棕色模块(n = 68,p = 1.07× 10 -24 )中显著富集。为了研究哪些共表达模块与 NASH 状态相关,作者将 eigengenes(代表每个模块表达谱的基因)的表达与 NASH 状态相关联。所有模块与 NASH 状态之间的关系显示在相关热图中,Y 轴对应基因组(模块),X 轴代表 NASH 状态(图 3E)。在 17 个共表达模块中,有 11 个 WGCNA 模块与 NASH 状态相关(P < 1.47 × 10 -3 ),这是在 Bonferroni 校正的基础上确定的。其中,9 个模块(青色、灰色60、绿松石色、品红色、紫色、浅青色、褐色、午夜蓝色和蓝色)与 NASH 疾病状态呈正相关,2 个模块(黑色和棕色)与 NASH 疾病状态呈负相关(图 3E)。
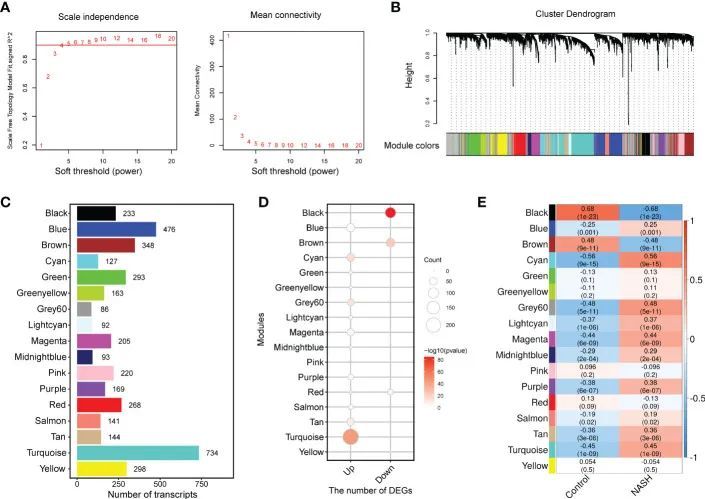
图3 WGCNA 网络和模块识别
4.感兴趣的共表达模块的功能表征
由于作者更关注在NASH组和对照组中表达不同的模块,因此作者比较了NASH样本和对照组在每个模块中的表达,并利用这些结果进一步评估模块是否与NASH状态相关。青色、灰色60和绿松石色模块在NASH中eigengenes上调,而黑色模块在NASH中表达较低(图4A)。为了研究共表达模块是否涵盖了与验证网络相关的信息,本研究利用 STRING 数据库中现有的蛋白质-蛋白质相互作用数据来检验检测到的模块的生物学特征。所有模块都显示出明显的相互作用富集(p < 0.01),因此表明本研究检测到的模块具有生物学相关性。此外,NASH 状态正相关的模块显示出更高的平均节点度(AND),尤其是 Turquoise 模块(AND = 22.4)。
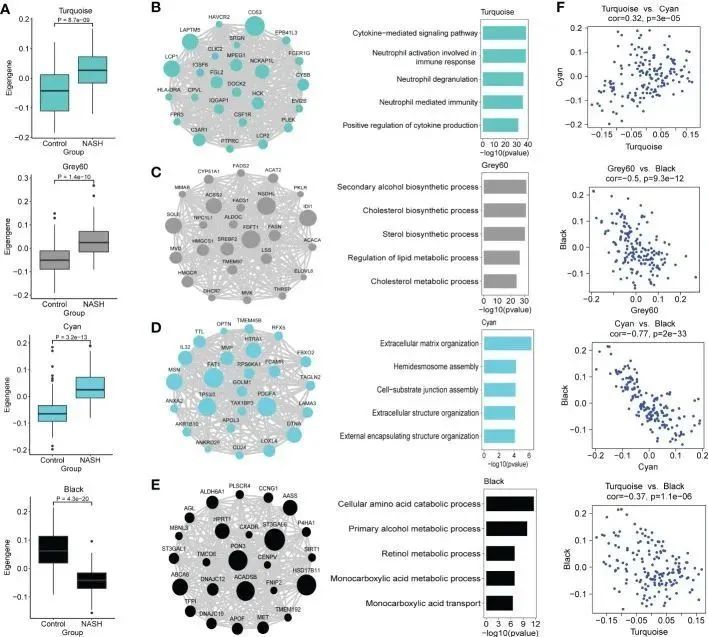
图4 WGCNA 鉴定出的感兴趣的共表达模块的功能特征
随后,作者利用 Enrichr 对 NASH 相关模块进行了 GO 和 KEGG 通路富集,以进一步研究其基因功能。各模块中的顶级生物过程和KEGG通路见表1。绿松石模块在NASH患者中上调,包含与免疫反应相关的关键基因(CD53、LAPTM5、LCP1、NCKAP1L、C3AR1和FGL2)(图4B),并富集了细胞因子介导的信号通路、参与免疫反应的中性粒细胞活化和中性粒细胞脱颗粒的GO类别(图4B)。Grey60模块中的FDFT1、NSDHL、IDI1、SQLE、ACSS2、SREBF2、HMGCR、FASN、LSS、ACAT2、FADS1、FADS2和ELOVL6等关键基因在NASH中上调(图4C),这些基因主要参与胆固醇和脂质代谢过程(图4C)。在青色模块中富集的大多数GO项主要与细胞外基质组织和细胞外基质结构组织相关(图4D),包括与纤维化相关的关键基因(PDGFA、LOXL4、MSN、LAMA3和AKR1B10)(图4D)。然而,黑色模块中富集的 GO 术语大多与细胞氨基酸分解代谢和初级酒精代谢过程有关(ACADSB、AASS 和 ALDH6A1)(图 4E)。作者接下来探讨了注释模块之间的基因关系。上调的免疫绿松石模块与纤维化相关的青色模块呈正相关(r = 0.32, p = 3.0 × 10 -5 )(图 4F),表明绿松石模块与驱动 NASH 纤维化的免疫反应有关,这证实了之前研究的结果(20)。有趣的是,青色、灰色 60 和绿松石模块与富含氨基酸代谢过程的黑色模块呈负相关(图 4F)。上调的纤维化模块 Cyan 与下调的富含代谢过程的 Black 模块之间存在高度负相关(r = -0.77,p = 2.0 × 10 -33 )(图 4F ),这表明氨基酸代谢紊乱可能参与了 NASH 的发病机制(39,40)。
5.模块保存分析表明,在免疫反应中存在与 NASH 相关的共表达模块功能
为了确定所发现的模块在另一个数据集中是否常见,作者研究了合并队列和最近发表的一个大型 NASH 数据集 GSE135251 ( 13) 之间的模块保留统计。具体而言,作者假定合并队列中的共表达模块为参考数据集,GSE135251 中的共表达模块为测试数据集。作者采用了 ( 22) 中描述的原理。Zsummary 得分大于 10 代表强保留模块,小于 2 表示非保留模块,而值在 2-10 之间则表示中度保留模块。作者绘制了 Zsummary 分数与 MergeCohort 模块大小的散点图(图 5A)。所有模块的 Zsummary 统计量都大于 2,表明所有模块都在 GSE135251 中得到了保留。保存率最低的是红色模块(Zsummary = 6.37)。尤其是合并队列模块绿松石(MergeCohort_Turquoise)的 Zsummary 保留分(Zsummary = 42.68)高于 40。为了更直观地反映每个共表达模块的保存情况,作者评估了 MergeCohort 和 GSE135251 的模块重叠情况(图 5B),发现 MergeCohort_Turquoise 与 GSE135251 模块 Turquoise(GSE135251_Turquoise)的重叠程度最高。此外,作者还发现在 MergeCohort_Turquoise 和 GSE135251_Turquoise 中重叠的 289 个基因的模块内连接性与 GSE135251_Turquoise 呈高度正相关(Spearman's correlation = 0.62, p = 1.3 × 10 -9 )(图 6A, B ),这表明这两个模块具有相似的共表达模式。
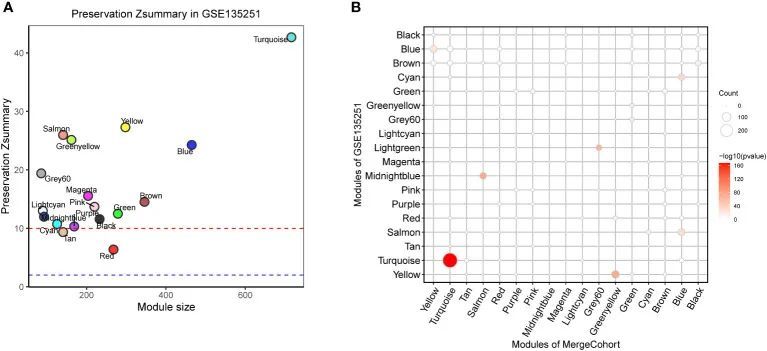
图5 GSE135251 数据集中 MergeCohort 的模块保存情况
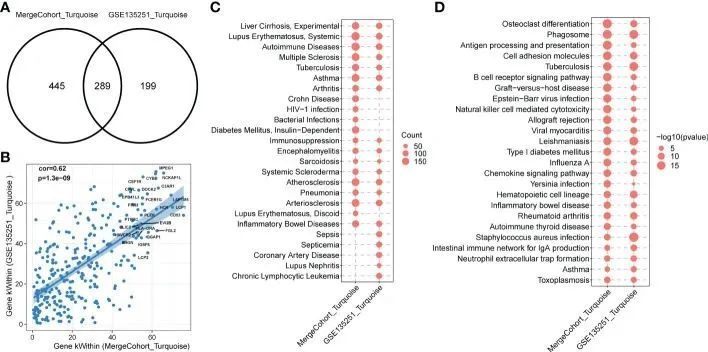
图6 MergeCohort_Turquoise 和 GSE135251_Turquoise 模块的功能富集
为了全面评估与 MergeCohort_Turquoise 和 GSE135251_Turquoise 相关的生物学功能,作者接下来计算了与 DisGeNET 数据库(33)中疾病相关基因集和 KEGG 通路基因集相关的基因富集的统计学意义。作者观察到,MergeCohort_Turquoise 和 GSE135251_Turquoise 中的基因被肝脏疾病相关基因集(肝硬化)和多种免疫疾病相关基因集(自身免疫性疾病、免疫抑制和炎症性肠病)显著富集(图 6C)。有趣的是,这两个模块在动脉粥样硬化和动脉硬化中也显著富集。值得注意的是,作者观察到,与 GSE135251_Turquoise 模块相似度最高的 MergeCohort_Turquoise 中的基因(734 个中有 289 个;超几何检验 p 值 = 5.33 × 10 -168 )(图 6A)在吞噬体、破骨细胞分化、细胞粘附分子、抗原处理和呈递、B 细胞受体信号通路中都有明显的富集(图 6D)。此外,MergeCohort_Turquoise 在 NASH 中上调,也是第三个最显著的模块,并显示出更多的统计学差异表达基因,734 个基因中有 233 个基因上调(折合变化 > 1.2;P < 0.05),没有一个基因显著下调(图 3D)。考虑到所有这些结果,作者将选择 MergeCohort 中的共表达绿松石模块进行进一步分析。
6.绿松石模块中枢基因的验证
NASH患者肝脏中的关键基因被上调。以 MergeCohort_Turquoise 模块为中心,作者首先探索了前 25 个具有高模内连接性(K.in)的关键基因,包括 CD53、LCP1、LAPTM5、NCKAP1L、C3AR1、PLEK、FCER1G、HLA-DRA 和 SRGN。这些关键基因的表达水平在本研究涉及的四个队列(GSE130970、GSE48452、GSE61260 和 GSE63067)中均呈上调趋势(图 7A),这表明这些关键基因可能在 NASH 的发生发展中起着基础性作用。图 7B 显示了这 25 个关键基因的 PPI 网络。

图7 MergeCohort_Turquoise 模块中关键基因的验证
关键基因与临床特征呈正相关。作者进一步研究了 GSE130970 中这 25 个关键基因的表达变化与组织学表型之间的关系(图 7C)。结果表明,25个关键基因均与非酒精性脂肪肝活动评分呈正相关,其中FPR3的相关性最高(r = 0.53, p = 1.49 × 10 -4 )。LCP1 基因是与脂肪变性等级(r = 0.46,p = 1.16 × 10 -3 )和小叶炎症等级(r = 0.32,p = 3.06 × 10 -2 )相关性最高的基因。此外,FPR3 与细胞学气球分级的相关性最大(r = 0.53,p = 1.82 × 10 -4 )。SRGN 是与纤维化分期最相关的基因(r = 0.35,p = 1.84 × 10 -2 )。此外,C3AR1 与所有临床参数都有显著相关性,尤其是与细胞学气球分级的相关性更高(r = 0.51,p = 2.94 × 10 -4 )。在胆碱缺乏L-氨基酸定义的高脂饮食(CDAHFD)小鼠NASH模型的肝脏中,关键基因被上调。此外,为了探索关键基因在小鼠中的意义,作者挖掘了公开的微阵列数据(GSE120977)(41),以验证上述基因的mRNA水平,但数据集中缺乏Hla-dra、Clic2和Fpr3基因。耐人寻味的是,与对照组相比,喂食 CDAHFD 食物 12 周的小鼠体内有几个中枢基因的表达量显著增加或呈上升趋势。例如,有14个基因,即Cd53、Laptm5、Nckap1l、C3ar1、Hck、Mpeg1、Cybb、Iqgap1、Dock2、Plek、Fcer1g、Igsf6、Ptprc和Havcr2,在喂食CDAHFD饲料的小鼠中强烈上调(图7D),支持了这些关键基因在小鼠NASH模型进展过程中也被激活的观点。
7.通过单细胞 RNA 序列分析鉴定细胞群对 NASH 相关 Turquoise 模块的贡献
为了研究在 MergeCohort_Turquoise 模块中发现的潜在关键基因在 NASH 进展过程中如何在特定细胞群中发生变化,作者使用来自健康和肝硬化肝脏样本的公开 scRNA-seq 数据进行了综合 scRNA-seq 分析。聚类显示了 17 个细胞群,包括 10 种不同的细胞类型(图 8A、B)。作者根据整合人类肝细胞图谱的发现和 SingleR 的注释分析,在 scRNA-seq 数据中识别出了内皮细胞、巨噬细胞、胆管细胞、NK 细胞、T 细胞、间质、树突状细胞、B 细胞、成纤维细胞和肝细胞。通过对肝组织进行 scRNA-seq 分析,分析了 MergeCohort_Turquoise 模块中前 25 个基因的表达模式。其中,CD53、LCP1、LAPTM5、PTPRC和SRGN等关键基因主要表达于不同的免疫细胞,如巨噬细胞、NK细胞、T细胞、树突状细胞和B细胞,而FGL2、HCK、MPEG1、CYBB、CSF1R、IGSF6、CPVL和HLA-DRA等关键基因则主要表达于巨噬细胞和树突状细胞。
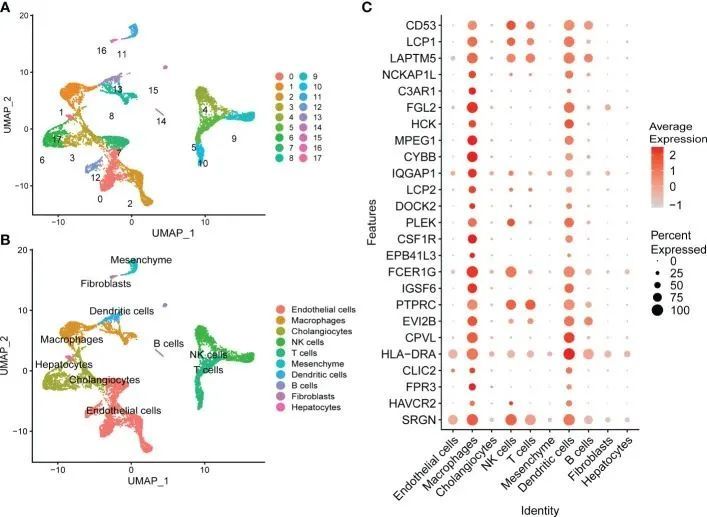
图8
8.识别调控绿松石模块的 TF
上述分析结果表明,MergeCohort_Turquoise 模块中的关键基因富集于免疫中。由于共表达基因往往受共同转录因子(TFs)的共调控,作者进一步利用 MergeCohort_Turquoise 和 GSE135251_Turquoise 模块中的基因进行了 TFs 富集分析(超几何检验),以获得基于 TRRUST 数据库 ( 34) 的关键调控基因。结果表明,NFKB1、SPI1、RELA、CIITA、HIVEP2、SP1、RFXANK、RFXAP、RFX5、IRF1 是 MergeCohort_Turquoise 模块中前 10 个显著富集的 TFs(图 9A)。此外,作者还采用了 ChEA3 数据库(35)来验证显著富集于 MergeCohort_Turquoise 模块基因的转录因子。结果,通过 ChEA3 分析,作者在 TRRUST 数据库中发现了 33 个对 MergeCohort_Turquoise 模块基因有意义的转录因子中的 27 个,另外 6 个转录因子是其靶标的一部分(表 S12)。作者还发现,NFKB1、SPI1、RELA、CIITA、SP1、RFXANK、RFXAP、RFX5、TRERF1、ELF1、STAT3、ERG、ETS1、ILF3、CEBPA、HDAC1 和 IRF8 是 MergeCohort_Turquoise 和 GSE135251_Turquoise 模块中显著富集的 TF(图 9A)。此外,与对照组相比,作者观察到在非酒精性脂肪肝和NASH中RFX5、ILF3、NFKB1、STAT3、ELF1、SPI1、ETS1和CEBPA的肝脏表达明显增加(P < 0.05)(图9B)。
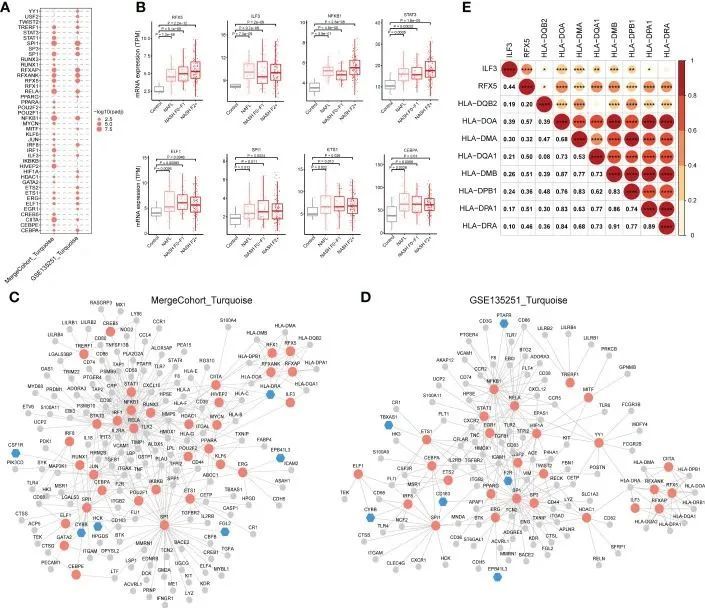
图9 NASH 相关模块中富集的转录因子与其靶基因之间的调控关系
接下来,作者为每个模块中富集的 TFs 和相关的靶基因构建了调控网络(图 9C、D)。作者观察到,RFX5和ILF3是主要在肝脏表达的重要转录因子,从轻度到晚期NASH均上调,它们调控参与抗原加工和通过MHC II类呈现外源肽抗原的基因的表达,包括HLA-DQB2、HLA-DOA、HLA-DMA、HLA-DQA1、HLA-DMB、HLA-DPB1、HLA-DPA1和HLA-DRA。值得注意的是,RFX5 和 ILF3 的基因表达与 MHCII 基因表达呈正相关(图 9E)。作者发现 41 个基因受 NFKB1 转录因子调控。众所周知,NFKB1 可调控细胞因子介导的信号通路(如 TNF、CXCL10、MMP9 和 TGFB1)和免疫反应(如 CD74、CD58、CD80 和 CD86)相关基因的表达(图 9C)。此外,STAT3 还调控伤口愈合过程中涉及炎症反应的基因的表达,包括 HMOX1、TIMP1、TGFB1 和 F2R。有趣的是,SPI1 调控参与免疫效应过程的基因(如 CTSG、CD68、IFIT3 和 IL18),包括 MergeCohort_Turquoise 模块中的关键基因(CYBB 和 HCK)。参与细胞活化的 SP1 调控基因(如 TIMP1、LTF、FGL2 和 LYZ)。
为了进一步分析 NASH 体外模型中关键基因和关键 TFs 的表达,作者检索了公开的 RNA-seq 数据(L02 肝细胞(PRJNA726826)和小鼠原代肝细胞(PRJNA726846)分别在棕榈酸和油酸(PAOA)处理 0h、12h 和 24h ( 42)),作者发现中枢基因(CD53 和 SRGN)和关键 TFs(NFKB1、ELF1 和 EST1)在 PAOA 处理的 L02 肝细胞中表达较高。此外,作者还观察到,在 PAOA 处理的小鼠原代肝细胞中,关键基因(Lcp1 和 Fcer1g)和关键 TF(Ilf3、stat3 和 Est1)的表达量增加。总之,作者研究中发现的这些TFs和靶基因为有兴趣对NASH的体外和体内机制和治疗方法进行临床前研究的研究人员或公司提供了一份很有前景的清单。
总结
总之,与以往局限于少数人类 NASH 转录组数据或关注影响 NASH 进展的单个基因的研究不同,作者的网络驱动策略产生了与 NASH 相关的模块、关键基因和关键转录因子的全面而公正的观点。特别是涉及免疫相关通路的 Turquoise 模块和调控因子,尤其是在 NASH 进展过程中协调抗原处理和呈递功能的转录因子 RFX5,值得进一步关注。本研究的主要局限性在于,所有结论均基于人类转录组数据,缺乏体外/体内疾病模型相关实验的验证。尽管如此,本研究还是为 NASH 预后和治疗靶点提供了有用的、新的分子候选者。