文章信息
原标题: Ternary NiMo-Bi liquid alloy catalyst for efficient hydrogen production from methane pyrolysis
中文标题:用于甲烷热解高效制氢的三元镍钼铋液态合金催化剂
作者:Luning Chen, Zhigang Song, Shuchen Zhang, Chung-Kai Chang, Yu-Chun Chuang, Xinxing Peng, Chaochao Dun, Jeffrey J. Urban, Jinghua Guo, Jeng-Lung Chen*, David Prendergast, Miquel Salmeron, Gabor A. Somorjai*, Ji Su*
通信单位:台湾同步辐射研究中心 加州大学伯克利分校 劳伦斯伯克利国家实验室
背景导读
氢(H2)作为一种优秀的清洁可再生能源,被视为最具前景的能源之一。2020年氢的总产量约9000万吨,然而,其大多数来自化石燃料,如天然气、石油和煤炭,这导致了大量的二氧化碳排放(约9亿吨)。水电解是一种绿色的氢生产技术,但高成本(5至6美元/kg)和高能耗(286 kJ/mol)导致目前水电解获得的氢仅占氢总产量的2 %。
甲烷(CH4)热解(MP)是另一种无二氧化碳排放的制氢方法,其理论能耗约为37.5 kJ/mol,但实际上需高温(>1000°C)促使甲烷活化,因此带来额外的能源消耗、设备成本以及不可避免的热耗散,适当的催化剂可以降低这些额外的成本。目前已有的催化剂包括固体催化剂(镍,钴,铁,铂和钯等过渡金属)和熔融液态催化剂(MLCs),前者具有较低的活化能,但容易因碳炼焦和芳烃污垢而失活,后者可以克服失活问题,但活化能较高,需要更高的温度才能活化甲烷。因此,如何开发出具有高催化活性的催化剂,使其工作温度适中,同时也具有良好的抗污垢和降解性能,一直以来是一个具有挑战性的问题。
文章简介
本文主要讲述了一种新型的液态合金催化剂,用于甲烷热解的高效产氢。研究人员通过在镍铋(Ni-Bi)液态合金中添加钼(Mo),制备了一种三元镍钼铋液态合金催化剂(LAC)。该催化剂在较低的活化能(81.2 kJ/mol)下表现出高效的甲烷热解活性,可以在450至800 ℃的范围内进行热解反应,在800 ℃下,催化剂表现出100%的氢气选择性和长达120小时的稳定性。这种催化剂具有高效、选择性和稳定性的特点,有望在甲烷热解反应中取得突破,并推动清洁能源的发展。

图1 用甲烷热解法制氢的方案。(A)不同催化剂的稳定性和活化能, (B)长时间甲烷热解反应后冷却产物的图像。

图2 催化数据。(A) 不同温度下的氢气产生速率,(B)在NiMo-Bi LAC上,甲烷热解的表观活化能(Ni/Mo的摩尔比为3:1)。(C) 在800°C下,不同组分的NiMo-Bi LAC的转化率、选择性(左)和H2产生速率(右),所有催化剂具有相同Ni含量。(D) 在800 ℃下,不同的液态合金催化剂的转化率和选择性(左)和H2生成速率(右)。(E) 在800 ℃和4 mol/min甲烷条件下, NiMo-Bi LAC(Ni/Mo的摩尔比为3:1)上的甲烷脱氢的长期稳定性测量。解法制氢的方案。(A)不同催化剂的稳定性和活化能, (B)长时间甲烷热解反应后冷却产物的图像。
光谱学研究结果验证了NiMo-Bi LAC中Ni和Mo相互作用的存在。Mo引入后,Ni和Mo之间的强相互作用调节了Ni的电子态,降低了Ni和Bi之间的相互作用,并导致Ni的负电荷减少,Mo在Ni-Bi体系中的溶解度增强。

图3 Ni-Bi和NiMo-Bi LAC中的Ni类型说明。(A)室温下Ni-Bi和NiMo-Bi LAC的2p x射线光电子能谱(XPS),(B)Ni-Bi和NiMo-Bi LAC在室温和工作温度下Ni的x射线吸收近边光谱(XANES)。
理论计算更为深入地给出了NiMo-Bi LAC的作用机理。
首先,研究人员模拟了Mo2二聚体和Mo团簇的溶解过程。在纯熔融态Bi中,Mo2二聚体的两个Mo原子的距离并没有因为其溶解度有限而改变,但在Ni的存在下,Mo-Mo的距离会增加。在Bi-Ni液态合金中的Mo团簇中也发现了类似的结果,这些结果表明,Ni-Mo相互作用使Mo在液体合金中均匀分散,而不是均匀聚集,从而提高了Mo的溶解度。这与实验观测相吻合。

图S16 (A) Ni-Bi液态合金和纯液态Bi中Mo2二聚体的Mo-Mo距离的变化。(B)Ni-Bi液态合金中的Mo团簇在溶解过程中不同截断半径(Rc)下的配位数的变化
紧接着,研究人员在Bi中嵌入了一个Ni原子,并在1500 K下进行了分子动力学模拟。(值得注意的是采取1500 K是为了加速模拟过程。)结果表明,Ni原子被Bi原子包围,Ni-Bi之间的强相互作用使得Ni处于Bi原子“笼”中;当在Mo引入后,Ni-Bi之间的相互作用减弱,Ni原子更容易与周围的Bi原子解离,因此更容易被甲烷分子作为催化活性位点。此外在图4(A)中,Ni原子的电荷降低也说明Mo的引入降低了Ni-Bi之间的相互作用。

图S17 Ni-Bi团簇在1500K的分子动力学模拟过程中的结构变化
图S18 NiMo-Bi团簇在1500K的分子动力学模拟过程中的结构变化
最后,研究人员在NiMo-Bi和Ni-Bi体系引入甲烷进行分子动力学模拟, 结果表明,Ni和Bi之间的强相互作用使得甲烷分子更难到达Ni活性位点,导致Ni-Bi体系的反应解离时间约为300飞秒(fs)。然而,Mo的引入减弱了这种笼效应,增加了Ni与甲烷相互作用的可能性,导致NiMo-Bi体系在约180 fs时开始解离。这些结果进一步支持了实验观察到的催化剂性能的提升。
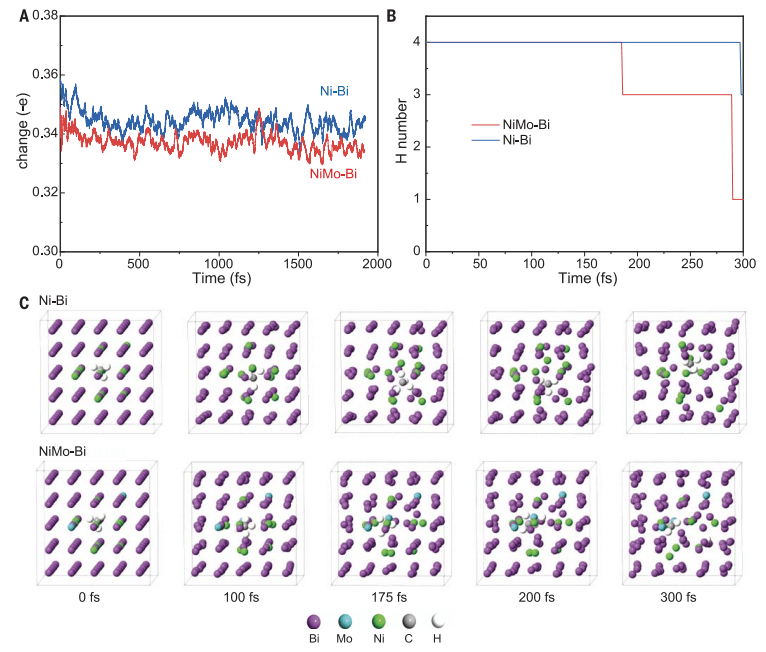
图4。分子动力学模拟。(A) 一个Ni原子上的平均电荷转移。(B)一个C原子周围的H原子数随时间演化。(C) 甲烷在Ni-Bi和NiMo-Bi催化剂中的解离过程。
本文计算数据均采用PWmat完成,本文理论部分由哈佛大学博士后宋志刚完成,是原文的共同一作。更多信息请查看原文:Chen, L., Song, Z., Zhang, S., Chang, C.-K., Chuang, Y.-C., Peng, X., et al. (2023). Ternary NiMo-Bi liquid alloy catalyst for efficient hydrogen production from methane pyrolysis. Science 381(6660), 857-861. doi: 10.1126/science.adh8872.