1、使用Trim Galore软件对两次数据进行质控,去掉20bp以下的reads
vim新建RNA_seq_script_1对CPIV3测序数据进行质控分析
#!/bin/bash
# 上面一行宣告这个script的语法使用bash语法,当程序被执行时,能够载入bash的相关环境配置文件。
# Program
# This program is used for RNA-seq data analysis.
# History
# 2023/07/13 zexing First release
# 设置变量${dir}为常用目录
dir=/home/customer/lizexing/projects/CPIV3/
# 使用fastqc软件对数据进行质控分析
# fastqc -t 8 -o ${dir}/fastqc_report/ ${dir}/raw_data/*.fq.gz
# 对数据利用Trim_galore去掉20bp以下的接头
trim_galore -q 8 --phred33 --stringency 3 --length 20 -e 0.1 -j 4 --paired \
${dir}/CleanData/C3_1.clean.fq \
${dir}/CleanData/C3_2.clean.fq \
-o ${dir}/clean_data/
后台运行RNA_seq_script_1:
nohup bash RNA_seq_script_1 > RNA_seq_script_1_log &
2、 使用Bowtie2软件对牛的基因组[bosTau9]构建索引
vim新建RNA_seq_script_2对牛的基因组构建索引
# 参数说明
# bowtie2 [options]* -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r> | --interleaved <i> | -b <bam>} [-S <sam>]
# <bt2-idx> Index filename prefix (minus trailing .X.bt2).
# NOTE: Bowtie 1 and Bowtie 2 indexes are not compatible.
# <m1> Files with #1 mates, paired with files in <m2>.
# Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
# <m2> Files with #2 mates, paired with files in <m1>.
# Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
# <r> Files with unpaired reads.
# Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
# <i> Files with interleaved paired-end FASTQ/FASTA reads
# Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
# <bam> Files are unaligned BAM sorted by read name.
# <sam> File for SAM output (default: stdout)
# <m1>, <m2>, <r> can be comma-separated lists (no whitespace) and can be specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
# bowtie2-build 基因组序列.fq 输出Index前缀 -p CPU数目
bowtie2-build GCF_002263795.2_ARS-UCD1.3_genomic.fna Bos -p 8
运行如下脚本:
nohup bash RNA_seq_script_2 > RNA_seq_script_2_log &
3、使用Tophat2软件对测序数据与牛的基因组进行比对
之前没有使用过Tophat2进行序列比对,这次因为第一次做病毒基因组从头组装,参考RNA病毒基因组组装指南、TopHat的使用以及输出文件安装、使用Tophat2软件进行比对。
(1)、先从Tophat官网下载相应的软件包进行安装;
(2)、使用Tophat需要提供bowtie软件提供的Index,官网已经提供了很多物种的Index及GTF格式文件,我自己还弄了半天;
(3)、tophat2报错:软件解压缩后,运行依赖于python2才可以运行,而现在的服务器默认的多为python3,看了网上很多办法,基本都是通过conda建立一个python2的环境后再运行Tophat2,我这边因为只能连接校园网,还没跟清华镜像联网,导致conda无法建立新环境(实属无奈),上网查到这个简便方法:
# 在tophat文件夹中打开tophat脚本
vim tophat
# 对该脚本的第一行进行python版本的指定
原命令为:#!/usr/bin/env python
新命令为:#!/usr/bin/env python2
# 仅添加了一个2的版本,省却了我很大的麻烦,必须记录、推广一下
(4)、通过help命令查看软件运行情况
(base) [customer@node01 tophat]$ bash tophat2 -h
Usage:
tophat [options] <bowtie_index> <reads1[,reads2,...]> [reads1[,reads2,...]] \
[quals1,[quals2,...]] [quals1[,quals2,...]]
(5)、按照默认参数对测序结果进行比对,命令如下:
#!/bin/bash
# 上面一行宣告这个script的语法使用bash语法,当程序被执行时,能够载入bash的相关环境配置文件。
# This program is used for RNA-seq data analysis.
# History
# 2023/07/13 zexing First release
# 设置变量${dir}为常用目录
# dir=/home/customer/lizexing/projects/CPIV3/
# tophat [options] <bowtie_index> <reads1[,reads2,...]> [reads1[,reads2,...]] [quals1,[quals2,...]] [quals1[,quals2,...]]
#-o 输出文件位置
#-p 几个CPU
#-G gtf文件
#index文件
#reads1
#reads2
tophat2 -o /home/customer/lizexing/projects/CPIV3/clean_data/mapped \
-p 8 \
-G /home/customer/lizexing/references/NCBI/Bostaurus/GCF_002263795.2.gtf \
/home/customer/lizexing/references/NCBI/Bostaurus/bt2_index/Bos \
/home/customer/lizexing/projects/CPIV3/clean_data/C3_1.clean_val_1.fq \
/home/customer/lizexing/projects/CPIV3/clean_data/C3_2.clean_val_2.fq
运行的时间比较长,我是利用nohup命令将其放到了后台运行。
# 软件运行记录如下:
[2023-07-14 13:32:23] Beginning TopHat run (v2.1.0)
-----------------------------------------------
[2023-07-14 13:32:23] Checking for Bowtie
Bowtie version: 2.4.2.0
[2023-07-14 13:32:23] Checking for Bowtie index files (genome)..
[2023-07-14 13:32:23] Checking for reference FASTA file
Warning: Could not find FASTA file /home/customer/lizexing/references/NCBI/Bostaurus/bt2_index/Bos.fa
[2023-07-14 13:32:23] Reconstituting reference FASTA file from Bowtie index
Executing: /home/customer/.conda/envs/RNA-ChIP-Seq/bin/bowtie2-inspect /home/customer/lizexing/references/NCBI/Bostaurus/bt2_index/Bos > /home/customer/lizexing/projects/CPIV3/tophat_out/tmp/Bos.fa
[2023-07-14 13:34:02] Generating SAM header for /home/customer/lizexing/references/NCBI/Bostaurus/bt2_index/Bos
[2023-07-14 13:34:04] Reading known junctions from GTF file
[2023-07-14 13:34:24] Preparing reads
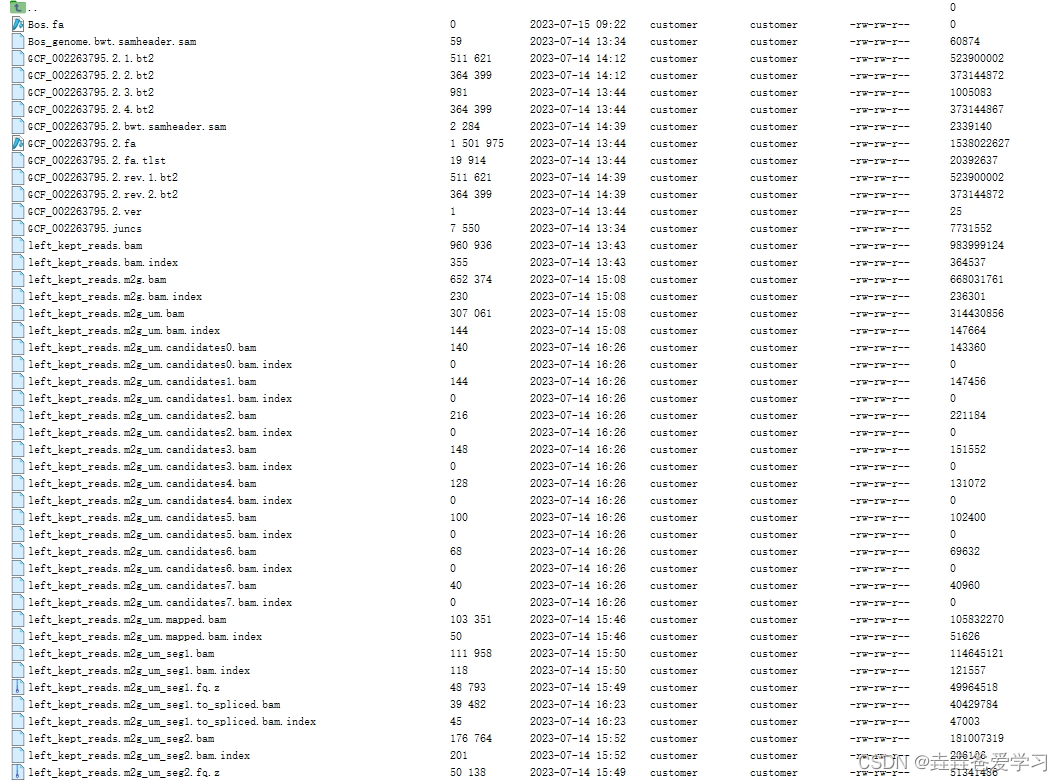
left reads: min. length=20, max. length=150, 15779194 kept reads (18 discarded)
right reads: min. length=20, max. length=150, 15779192 kept reads (20 discarded)
[2023-07-14 13:43:14] Building transcriptome data files /home/customer/lizexing/projects/CPIV3/tophat_out/tmp/GCF_002263795.2
[2023-07-14 13:44:09] Building Bowtie index from GCF_002263795.2.fa
[2023-07-14 14:39:34] Mapping left_kept_reads to transcriptome GCF_002263795.2 with Bowtie2
[2023-07-14 15:08:02] Mapping right_kept_reads to transcriptome GCF_002263795.2 with Bowtie2
[2023-07-14 15:39:45] Resuming TopHat pipeline with unmapped reads
[2023-07-14 15:39:46] Mapping left_kept_reads.m2g_um to genome Bos with Bowtie2
[2023-07-14 15:49:49] Mapping left_kept_reads.m2g_um_seg1 to genome Bos with Bowtie2 (1/6)
[2023-07-14 15:50:58] Mapping left_kept_reads.m2g_um_seg2 to genome Bos with Bowtie2 (2/6)
[2023-07-14 15:52:20] Mapping left_kept_reads.m2g_um_seg3 to genome Bos with Bowtie2 (3/6)
[2023-07-14 15:53:30] Mapping left_kept_reads.m2g_um_seg4 to genome Bos with Bowtie2 (4/6)
[2023-07-14 15:54:42] Mapping left_kept_reads.m2g_um_seg5 to genome Bos with Bowtie2 (5/6)
[2023-07-14 15:56:11] Mapping left_kept_reads.m2g_um_seg6 to genome Bos with Bowtie2 (6/6)
[2023-07-14 15:57:39] Mapping right_kept_reads.m2g_um to genome Bos with Bowtie2
[2023-07-14 16:09:56] Mapping right_kept_reads.m2g_um_seg1 to genome Bos with Bowtie2 (1/6)
[2023-07-14 16:11:08] Mapping right_kept_reads.m2g_um_seg2 to genome Bos with Bowtie2 (2/6)
[2023-07-14 16:12:26] Mapping right_kept_reads.m2g_um_seg3 to genome Bos with Bowtie2 (3/6)
[2023-07-14 16:13:45] Mapping right_kept_reads.m2g_um_seg4 to genome Bos with Bowtie2 (4/6)
[2023-07-14 16:15:08] Mapping right_kept_reads.m2g_um_seg5 to genome Bos with Bowtie2 (5/6)
[2023-07-14 16:16:38] Mapping right_kept_reads.m2g_um_seg6 to genome Bos with Bowtie2 (6/6)
[2023-07-14 16:18:19] Searching for junctions via segment mapping
[2023-07-14 16:21:49] Retrieving sequences for splices
[2023-07-14 16:22:57] Indexing splices
Building a SMALL index
[2023-07-14 16:23:10] Mapping left_kept_reads.m2g_um_seg1 to genome segment_juncs with Bowtie2 (1/6)
[2023-07-14 16:23:34] Mapping left_kept_reads.m2g_um_seg2 to genome segment_juncs with Bowtie2 (2/6)
[2023-07-14 16:23:55] Mapping left_kept_reads.m2g_um_seg3 to genome segment_juncs with Bowtie2 (3/6)
[2023-07-14 16:24:16] Mapping left_kept_reads.m2g_um_seg4 to genome segment_juncs with Bowtie2 (4/6)
[2023-07-14 16:24:36] Mapping left_kept_reads.m2g_um_seg5 to genome segment_juncs with Bowtie2 (5/6)
[2023-07-14 16:25:00] Mapping left_kept_reads.m2g_um_seg6 to genome segment_juncs with Bowtie2 (6/6)
[2023-07-14 16:25:23] Joining segment hits
[FAILED]
Error running 'long_spanning_reads':Warning: 5737921 malformed closure
通过检查记录发现,首先对测序结果比对了转录本、基因组信息,然后对间隔片段进行比较,其中左侧的reads比对了间隔片段,右侧的reads比对间隔片段是报错了,然后程序就终止了。
(base) [customer@node01 tophat_out]$ cat prep_reads.info
left_min_read_len=20
left_max_read_len=150
left_reads_in =15779212
left_reads_out=15779194
right_min_read_len=20
right_max_read_len=150
right_reads_in =15779212
right_reads_out=15779192
因为第一次使用tophat2软件,不了解最终输出结果都有哪些,怀疑是程序中断未成功,故不再后续操作。
4、 使用STAR软件对牛的基因组[bosTau9]构建索引
vim新建RNA_seq_script_3对牛的基因组构建索引
# 参数说明
--runThreadN是指你要用几个cpu来运行;
--genomeDir构建索引输出文件的目录;
--genomeFastaFiles你的基因组fasta文件所在的目录
--limitGenomeGenerateRAM 43749387189 STAR消耗内存太大,输入限制内存数目防止出错,感谢孙小雨帮忙
STAR --runMode genomeGenerate --runThreadN 8 --limitGenomeGenerateRAM 82424365322 \
--genomeDir /home/customer/lizexing/references/NCBI/Bostaurus/star_index/ \
--genomeFastaFiles /home/customer/lizexing/references/NCBI/Bostaurus/GCF_002263795.2_ARS-UCD1.3_genomic.fna
运行如下脚本:
nohup bash RNA_seq_script_3 > RNA_seq_script_3_log &
5、使用STAR软件对测序数据与牛的基因组进行比对
vim新建RNA_seq_script_4对2023_07_13测序数据进行处理
#!/bin/bash
# 上面一行宣告这个script的语法使用bash语法,当程序被执行时,能够载入bash的相关环境配置文件。
# This program is used for RNA-seq data analysis.
# History
# 2023/07/13 zexing First release
# 设置变量${dir}为常用目录
dir=/home/customer/lizexing/projects/CPIV3/
STAR --runThreadN 8 --runMode alignReads --twopassMode Basic --outSAMtype BAM Unsorted \
--sjdbGTFfile /home/customer/lizexing/references/NCBI/Bostaurus/GCF_002263795.2.gtf \
--genomeDir /home/customer/lizexing/references/NCBI/Bostaurus/star_index/ \
--readFilesIn ${dir}/clean_data/C3_1_clean_val_1.fq ${dir}/clean_data/C3_2_clean_val_2.fq \
--outFileNamePrefix ${dir}/clean_data/Mapped/mapped-val \
--outReadsUnmapped Fastx
后台运行RNA_seq_script_4:
nohup bash RNA_seq_script_4 > RNA_seq_script_4_log &
查看运行日志结果如下:
(base) [customer@node01 Mapped]$ cat mapped-valLog.final.out
Started job on | Jul 13 16:26:23
Started mapping on | Jul 13 16:36:21
Finished on | Jul 13 16:42:24
Mapping speed, Million of reads per hour | 156.49
Number of input reads | 15779212
Average input read length | 299
UNIQUE READS:
Uniquely mapped reads number | 11785614
Uniquely mapped reads % | 74.69%
Average mapped length | 293.87
Number of splices: Total | 3037863
Number of splices: Annotated (sjdb) | 3014288
Number of splices: GT/AG | 2992015
Number of splices: GC/AG | 22366
Number of splices: AT/AC | 2758
Number of splices: Non-canonical | 20724
Mismatch rate per base, % | 0.34%
Deletion rate per base | 0.02%
Deletion average length | 1.25
Insertion rate per base | 0.01%
Insertion average length | 1.29
MULTI-MAPPING READS:
Number of reads mapped to multiple loci | 297690
% of reads mapped to multiple loci | 1.89%
Number of reads mapped to too many loci | 1235
% of reads mapped to too many loci | 0.01%
UNMAPPED READS:
Number of reads unmapped: too many mismatches | 0
% of reads unmapped: too many mismatches | 0.00%
Number of reads unmapped: too short | 3687419
% of reads unmapped: too short | 23.37%
Number of reads unmapped: other | 7254
% of reads unmapped: other | 0.05%
CHIMERIC READS:
Number of chimeric reads | 0
% of chimeric reads | 0.00%
检查发现比对上去的包括75%以上,同时输出的文件中有一个bam文件存储比对结果

6、使用samtools view软件将比对到基因组的bam文件转换为sam文件
参考文章:RNA病毒基因组组装指南
因为后边的python脚本需要输入sam文件,因此利用samtools view命令将bam文件转换为sam文件,如下:
[customer@node01 Mapped]$ samtools view -h -@ 4 -o mapped-valAligned.out.sam mapped-valAligned.out.bam
7、使用py脚本对测序数据进行处理
参考文章:RNA病毒基因组组装指南
“SAM文件的第一行的reads的名称和原来fastq文件夹里的reads名称是相同的,所以根据reads的名称就可以把SAM文件和原始的fastq文件关联起来。”
“知道了宿主来源的reads的名字之后,就可以在原始的fastq文件里查找并且删除了,对于每一个宿主来源的reads名称,我们可以遍历整个fastq文件来查找匹配到的reads然后把它删除(如果是paired-end reads的话就需要同时把两个fastq文件里相对应的reads都删掉),但是这样做会很慢,操作次数基本上是两个集合元素个数的乘积。所以可以先对两个reads名字的集合进行排序,因为匹配到的reads的名字的集合一定是所有的reads名字集合的一个子集,**排序之后,我们只需要将总reads集合里的每一个元素始终和小集合里第一个元素进行比较,如果发现相同,就把小集合里的第一个元素删掉,后面的推上来。**这样的话就可以在遍历一遍reads的情况下标记下所有map上去的reads,计算量就只是落在对两个集合排序上了,而排序所需要的操作次数比之前挨个查找的方法减少了几千倍。这个python小程序可以读取两个paired-end fastq文件(测序后处理掉接头蛋白的序列)和一个SAM文件(比对到基因组上的序列),然后用这种方式输出两个去除宿主来源之后的paired-end fastq文件。”
由github下载相应的脚本,运行命令如下:
# 该python脚本使用的是python2进行编译,所以需要使用python2进行运行
# 该脚本的使用方法如下:
usage: remove_mapped_pairdreads.py [-h] [-o OUTPUTDIR] read1 read2 input_sam
# 具体操作命令如下:
(RNA-ChIP-Seq) [customer@node01 Remove_mapped]$ python2 remove_mapped_pairdreads.py C3_1.clean_val_1.fq C3_2.clean_val_2.fq mapped-valAligned.out.sam
8、使用Velvet软件对测序结果进行从头组装
参考文章:velvet软件进行基因组组装、序列拼接 - Velvet
(1)、激活软件
从GitHub网站下载Velvet软件包后,在服务器解压缩。默认情况下,velvet文件夹中无可执行软件。利用make命令执行后才产生可执行软件,编译完成后,会生成两个可执行文件:velveth、velvetg
make 'MAXKMERLENGTH=127'
# 运行结果如下
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/shortReadPairs.c -o obj/shortReadPairs.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/locallyCorrectedGraph.c -o obj/locallyCorrectedGraph.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/graphReConstruction.c -o obj/graphReConstruction.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/roadMap.c -o obj/roadMap.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/preGraph.c -o obj/preGraph.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/preGraphConstruction.c -o obj/preGraphConstruction.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/concatenatedPreGraph.c -o obj/concatenatedPreGraph.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/readCoherentGraph.c -o obj/readCoherentGraph.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/utility.c -o obj/utility.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/kmer.c -o obj/kmer.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/scaffold.c -o obj/scaffold.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/kmerOccurenceTable.c -o obj/kmerOccurenceTable.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/allocArray.c -o obj/allocArray.o
gcc -Wall -m64 -O3 -D MAXKMERLENGTH=127 -D CATEGORIES=2 -c src/autoOpen.c -o obj/autoOpen.o
MAXKMERLENGTH=127: 默认情况下的最大的Kmer长度31。(k-mers一般选择17即可,对于高度重复基因组或者基因组过大,可以选择19甚至31也行。但不是越大越好,kmer越大,越耗内存,而且如果一条reads里有一个错误位点,越大的k-mers就会导致包含这个错误位点的k-mers个数增多); 如果想要只会更大长度的kmer,在编译时需要设置MAXKMERLENGTH的值。
(2)、软件自检
进入软件中的Test文件夹,运行run-tests.sh脚本对软件进行自检,结果如下:
(RNA-ChIP-Seq) [customer@node01 tests]$ bash run-tests.sh
[五 7月 14 14时56分24秒 2023] Running Velvet Test Suite
[五 7月 14 14时56分24秒 2023] Found ../velveth, ok
[五 7月 14 14时56分24秒 2023] Found ../velvetg, ok
[五 7月 14 14时56分24秒 2023] Found Sequences.31, ok
[五 7月 14 14时56分24秒 2023] Found Roadmaps.31, ok
[五 7月 14 14时56分24秒 2023] Found read1.fq.gz, ok
[五 7月 14 14时56分24秒 2023] Found read2.fq.gz, ok
[五 7月 14 14时56分24秒 2023] Found reads.fq.gz, ok
[五 7月 14 14时56分24秒 2023] Found read1.fa.gz, ok
[五 7月 14 14时56分24秒 2023] Found read2.fa.gz, ok
[五 7月 14 14时56分24秒 2023] Found reads.fa.gz, ok
[五 7月 14 14时56分24秒 2023] making test folder: velvet.test.oPQ3Lk
[五 7月 14 14时56分24秒 2023] running test 1 of 5: fasta_eq_fastq_ascii.t
[五 7月 14 14时56分26秒 2023] ok
[五 7月 14 14时56分26秒 2023] ok
[五 7月 14 14时56分26秒 2023] ok
[五 7月 14 14时56分26秒 2023] ok
[五 7月 14 14时56分26秒 2023] passed test 1
[五 7月 14 14时56分26秒 2023] running test 2 of 5: fasta_eq_fastq_binary.t
[五 7月 14 14时56分27秒 2023] ok
[五 7月 14 14时56分27秒 2023] ok
[五 7月 14 14时56分27秒 2023] passed test 2
[五 7月 14 14时56分27秒 2023] running test 3 of 5: fmt_auto_mode.t
[五 7月 14 14时56分29秒 2023] ok
[五 7月 14 14时56分29秒 2023] ok
[五 7月 14 14时56分29秒 2023] passed test 3
[五 7月 14 14时56分29秒 2023] running test 4 of 5: interleaved_eq_separate.t
[五 7月 14 14时56分32秒 2023] ok
[五 7月 14 14时56分32秒 2023] ok
[五 7月 14 14时56分32秒 2023] passed test 4
[五 7月 14 14时56分32秒 2023] running test 5 of 5: mismatched_separates.t
[五 7月 14 14时56分32秒 2023] ok
[五 7月 14 14时56分32秒 2023] ok
[五 7月 14 14时56分32秒 2023] passed test 5
[五 7月 14 14时56分32秒 2023] removing test folder: velvet.test.oPQ3Lk
[五 7月 14 14时56分32秒 2023] passed all 5 tests
[五 7月 14 14时56分32秒 2023] hooray!
(3)、通过make命令设置其他参数
# 输入 10 groups of short reads。根据原始数据相应增减该值的大小;值越大,耗内存越大。
make 'CATEGORIES=10'
# 超过 2.2G 的reads用于组装基因组的时候,需要设置该值。
make 'BIGASSEMBLY=1'
# 当contigs长度超过 32kb 长的时候,需要设置该值。
make 'LONGSEQUENCES=1'
# 多线程运行。需要设置环境变量 OMP_NUM_THREADS 和 OMP_THREAD_LIMIT。最多为 OMP_NUM_THREADS+1 或 OMP_THREAD_LIMIT 个线程.
make 'OPENMP=1'
# velvet默认使用系统自带的zlib,如果系统没有zlib,则需要加入该参数来使用velvet源码包中的zlib.
make 'BUNDLEDZLIB=1'






![[QT编程系列-11]:C++图形用户界面编程,QT框架快速入门培训 - 5- QT主要控件与自定义控件](https://img-blog.csdnimg.cn/b3b4d4442f3e4ccfa859a1600abf64a7.png)