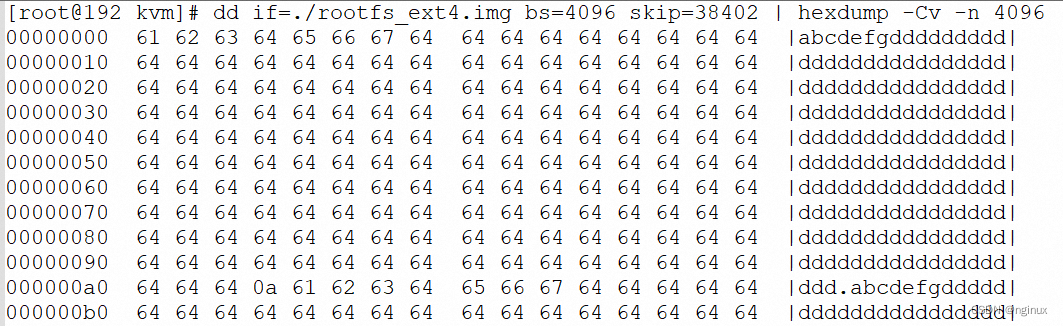
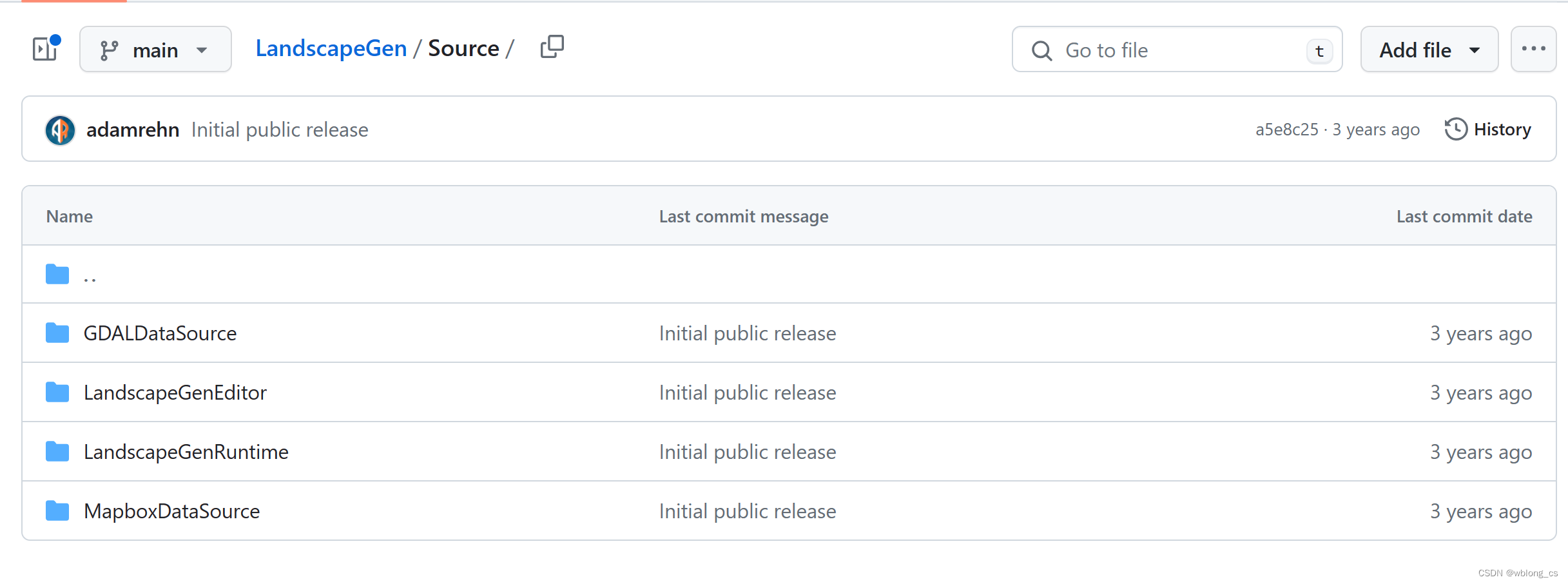
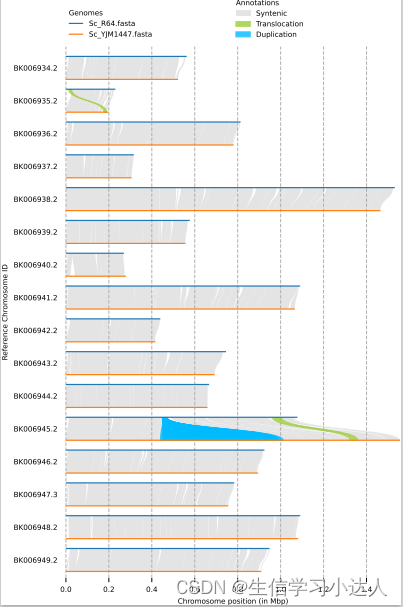
为了对 syri.out (assembly的变异检测结果)进行可视化处理,本人选择了plotsr软件对其基因组重排现象进行可视化:
(base) [hgzhong@head01 08.assembly_calling]$ plotsr --sr syri.out --genomes ../data/Sc_R64.fasta --genomes ../data/Sc_YJM1447.fasta -H 8 -W 5
2023-07-14 21:41:50,978 - Plotsr - INFO - Starting
Traceback (most recent call last):
File "/data/home/hgzhong/miniconda3/bin/plotsr", line 6, in <module>
main(sys.argv[1:])
File "/data/home/hgzhong/miniconda3/lib/python3.10/site-packages/plotsr/scripts/plotsr.py", line 334, in main
plotsr(args)
File "/data/home/hgzhong/miniconda3/lib/python3.10/site-packages/plotsr/scripts/plotsr.py", line 171, in plotsr
chrlengths, genomes = validalign2fasta(alignments, args.genomes.name)
File "/data/home/hgzhong/miniconda3/lib/python3.10/site-packages/plotsr/scripts/func.py", line 1017, in validalign2fasta
raise ImportError("Incomplete genomic information.\nExpected format for the genome file:\npath_to_genome1\tgenome1_id\ttags\npath_to_genome2\tgenome2_id\ttags\n\nMake sure that the columns are separated by tabs (and not spaces).")
ImportError: Incomplete genomic information.
Expected format for the genome file:
path_to_genome1 genome1_id tags
path_to_genome2 genome2_id tags出现“ImportError: Incomplete genomic information” 报错,
## 需准备两个物种的基因组列表
python3 $PATH_TO_PLOTSR --sr syri.out --genomes genomes
cat genomes
/home A.chr.fa refgenome
/home B.chr.fa refgenome
but the error appear.
ImportError: Incomplete genomic information.
Expected format for the genome file:
path_to_genome1 genome1_id tags
path_to_genome2 genome2_id tags
Make sure that the columns are separated by tabs (and not spaces).
so, what should be the format of the input file --genomes
Could you please guide if there is any solution, how to fix theproblem?仍然报错,显示genomes.txt的分割符应为 tab 而非空格
plotsr --sr syri.out --genomes genomes -H 8 -W 5参数:
--sr : 指定syri.out文件
-- genomes: 两个物种的基因组列表,该文件含有2列,第一列为物种基因组的路径,第二列为 genome_id。注意此2列的分隔符为tab(非空格)--参考网站 报错网址
-H: plot 图的高度
-W: plot 图的宽度

![]()
问题成功解决,最终获得plotsr.pdf文件。
![Unity Shader - SV_POSITION 和 TEXCOORD[N] 的varying 在 fragment shader 中输出的区别](https://img-blog.csdnimg.cn/b25bef8b30844ed5815298fafa04e3d2.png)