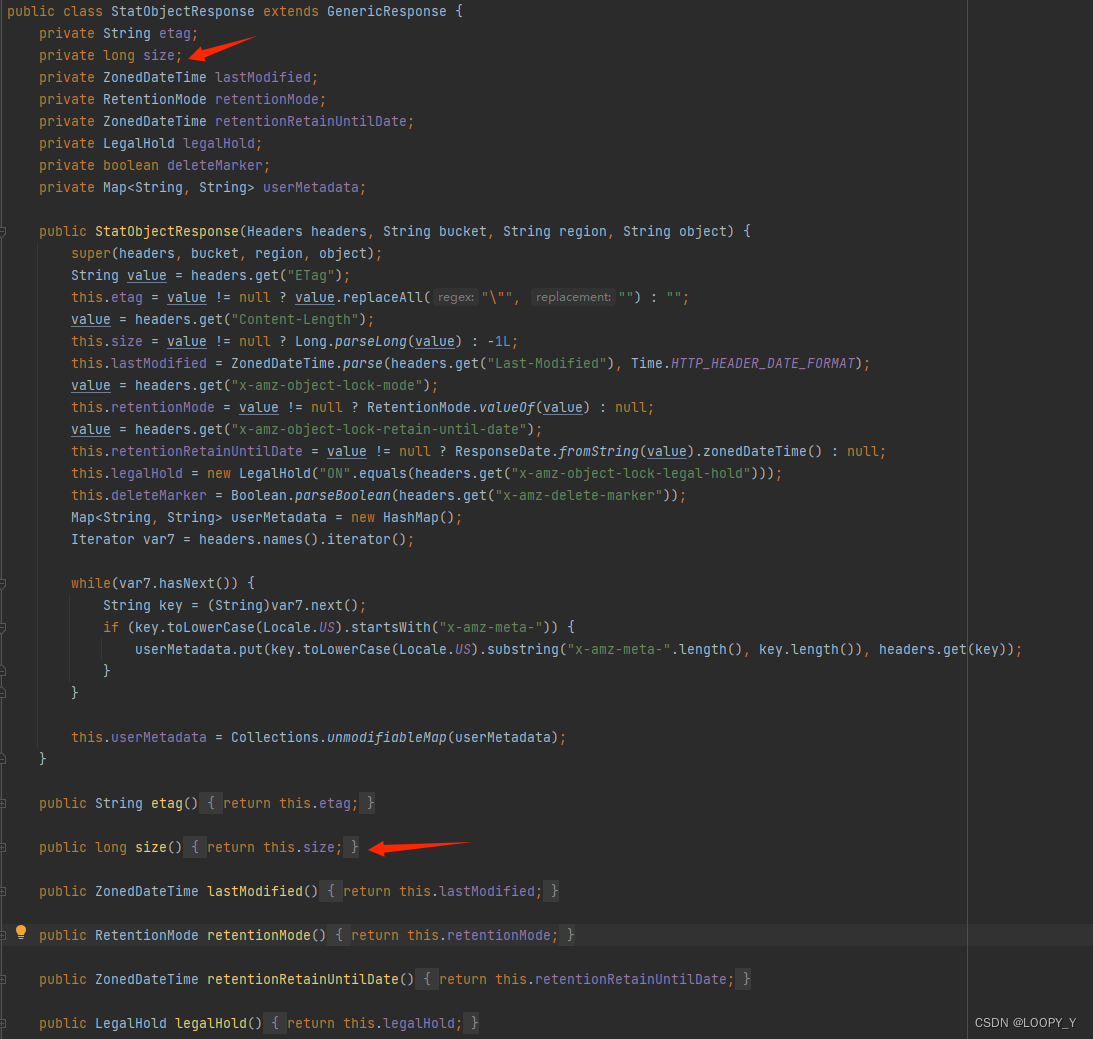
github:地址
文章目录
- Rdkit|最大公共子结构
- `rdFMCS.FindMCS`
- 参数
- `bondCompare`
- `atomCompare`
- `matchValences`
- `ringMatchesRingOnly`
- `completeRingsOnly`
- matchChiralTag
- 高亮分子的不同子结构
- 参考
Rdkit|最大公共子结构
rdFMCS.FindMCS
mols: 分子对象maximizeBonds一个结构由原子和键组成。默认True情况下,MCS查找的目标是让键的数量最大化,相当于让环的数量最大化。最大化键的数量时,会使环的数量最大化,但也可能出现两个小环键的数量不如一个大环多的情况。可以通过将maximizeBonds参数设为False来取消设置。threshold:线程数,默认:1timeout:超时时间,默认:3600,MCS算法会穷尽每一种可能,寻找到一个最大的公共子结构。一般情况下说,该算法几秒内就能完成,但是有时也会花很长时间甚至花几分钟。这时,可以使用timeout参数来规定最大搜索时长,到达指定时长仍没有搜索完时,会返回现有的最优结果,并将canceled属性设置为True。该参数单位为秒。verbose:输出日志,默认:FalsematchValences:即忽略化合价信息,默认False。假设要考虑化合价信息,例如不想让一个3价氮与一个5价氮匹配上,那就可以将该参数改为True。ringMatchesRingOnly: 默认为False,这种情况下,线性的碳链可能会匹配上一个环。如果只想让环相互匹配,可以将该参数设置为True。completeRingsOnly:如果只想要能够完整匹配上的环,而不想要匹配一半的环,可以将该参数设置为True。默认False。matchChiralTag: 是否匹配立体化学。默认忽略立体化学:False。如果matchChiralTag为 True,则算法有两处变化:- 具有指定手性的原子只能匹配其他具有指定手性的原子
- 如果 MCS 在指定手性的原子周围包含至少三个原子,则所有分子中的手性必须匹配。
atomCompare:原子匹配方式CompareElements:默认值,原子类型一样才算匹配。CompareIsotopes:同位素一样才算匹配(根据同位素标签的值。这个值可以由用户自己定义)。CompareAny: 表示任意原子之间都能匹配(找公共骨架)。
bondCompare: 键匹配方式CompareOrderExact:当且仅当键的类型完全一致才相等(芳香键和双键是不相等的)。CompareOrder:默认值为该方法。允许单键和芳香键相互匹配。CompareAny:任意键之间都可以匹配
ringCompare:环匹配方式seedSmarts: 随机种子
该函数返回一个MCSResult实例,实例含有以下属性:
MCSResult.queryMol: 搜索到结构的mol对象MCSResult.numAtoms: 搜索到结构的原子数MCSResult.numBonds: 搜索到结构的键数量MCSResult.smartsString: 搜索到结构的smartsMCSResult.canceled: 搜索是否超时

from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import rdFMCS
from rdkit.Chem import Draw
参数
bondCompare
mols = [Chem.MolFromSmiles('c1ccccc1'),Chem.MolFromSmiles('C1CCCC=C1')]
results = []
res = rdFMCS.FindMCS(mols)
print(res.smartsString)
results.append(Chem.MolFromSmarts(res.smartsString))
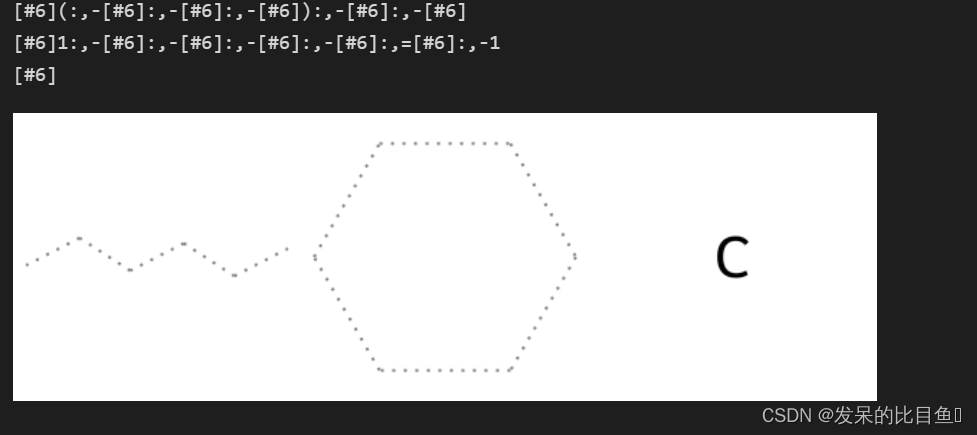
res = rdFMCS.FindMCS(mols, bondCompare=rdFMCS.BondCompare.CompareAny)
print(res.smartsString)
results.append(Chem.MolFromSmarts(res.smartsString))
res = rdFMCS.FindMCS(mols, bondCompare=rdFMCS.BondCompare.CompareOrderExact)
print(res.smartsString) # 结果为空
results.append(Chem.MolFromSmarts(res.smartsString))
Draw.MolsToImage(results)
atomCompare
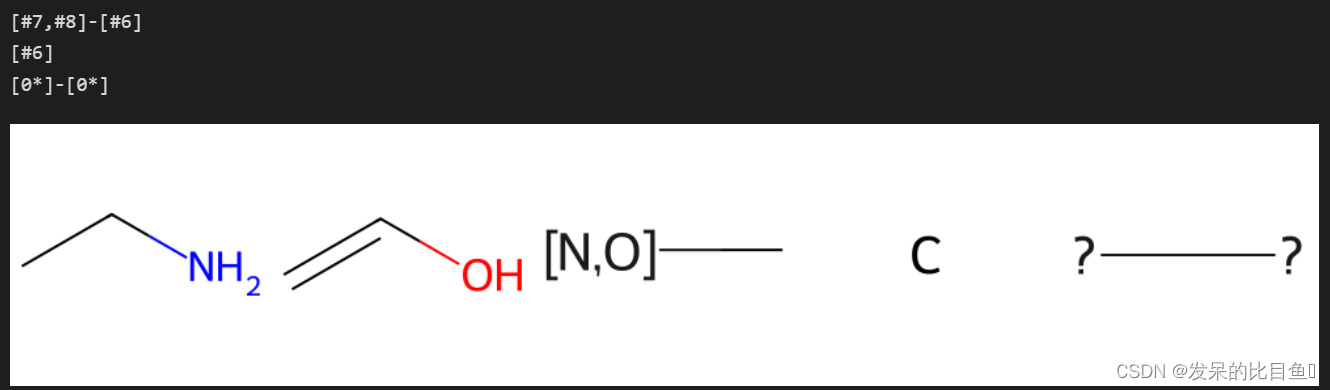
mols = (Chem.MolFromSmiles('NCC'),Chem.MolFromSmiles('OC=C'))
results = [Chem.MolFromSmiles('NCC'), Chem.MolFromSmiles('OC=C')]
res = rdFMCS.FindMCS(mols, atomCompare=rdFMCS.AtomCompare.CompareAny)
print(res.smartsString)
results.append(Chem.MolFromSmarts(res.smartsString))
res = rdFMCS.FindMCS(mols, atomCompare=rdFMCS.AtomCompare.CompareElements)
print(res.smartsString)
results.append(Chem.MolFromSmarts(res.smartsString))
res = rdFMCS.FindMCS(mols, atomCompare=rdFMCS.AtomCompare.CompareIsotopes)
print(res.smartsString)
results.append(Chem.MolFromSmarts(res.smartsString))
Draw.MolsToImage(results)
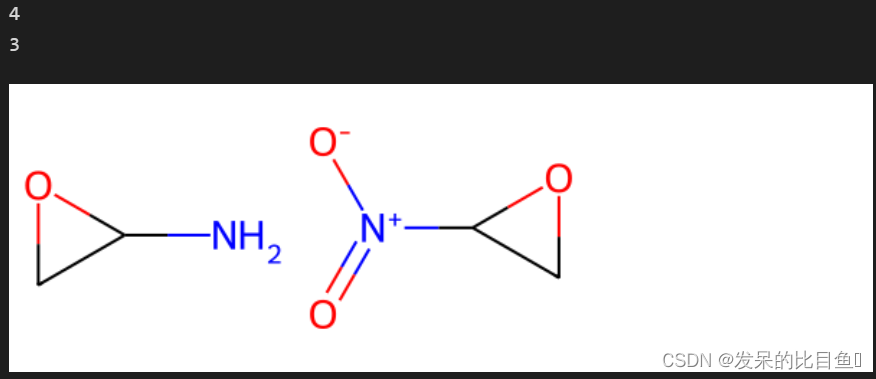
matchValences
mols = (Chem.MolFromSmiles('NC1OC1'),Chem.MolFromSmiles('C1OC1[N+](=O)[O-]'))
print(rdFMCS.FindMCS(mols).numAtoms)
print(rdFMCS.FindMCS(mols, matchValences=True).numBonds)
Draw.MolsToGridImage(mols)
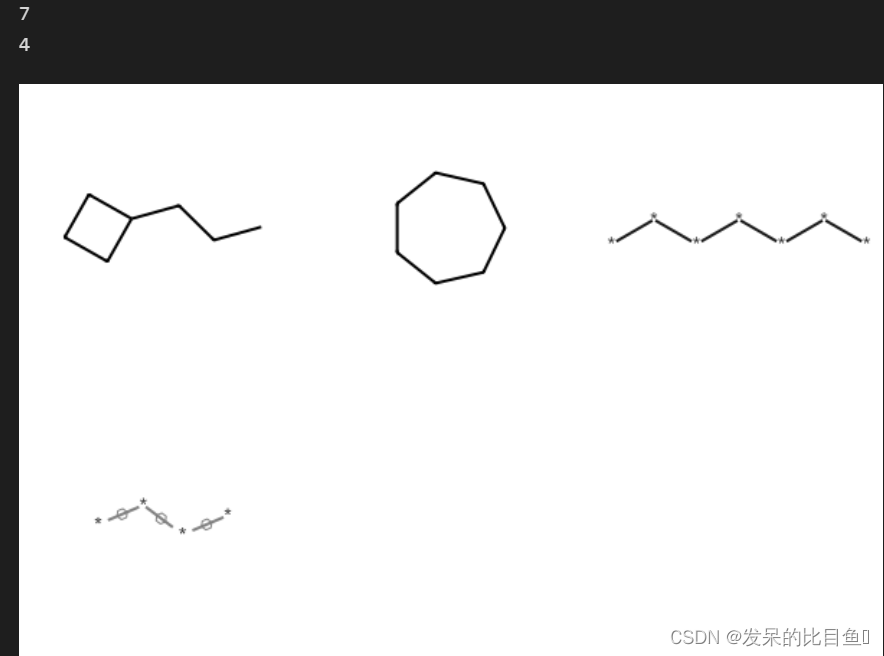
ringMatchesRingOnly
mols = [Chem.MolFromSmiles("C1CCC1CCC"), Chem.MolFromSmiles("C1CCCCCC1")]
res1 = rdFMCS.FindMCS(mols)
print(res1.numAtoms)
res2 = rdFMCS.FindMCS(mols, ringMatchesRingOnly=True)
print(res2.numAtoms)
mols += [res1.queryMol, res2.queryMol]
Draw.MolsToGridImage(mols)
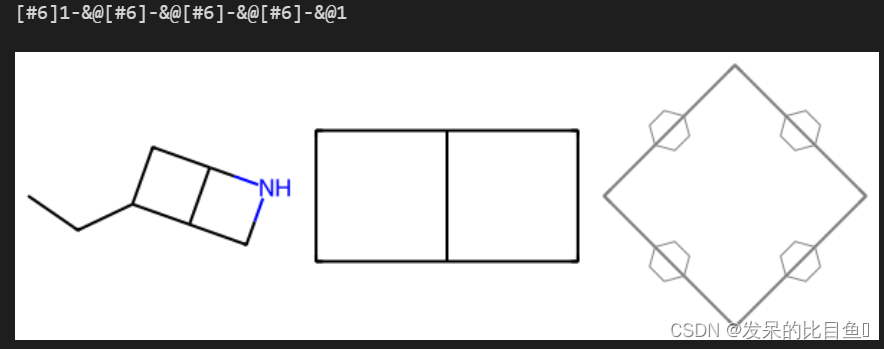
completeRingsOnly
mols = [Chem.MolFromSmiles("CCC1CC2C1CN2"), Chem.MolFromSmiles("C1CC2C1CC2")]
res = rdFMCS.FindMCS(mols, completeRingsOnly=True).smartsString
print(res)
Draw.MolsToImage(mols+[Chem.MolFromSmarts(res)])
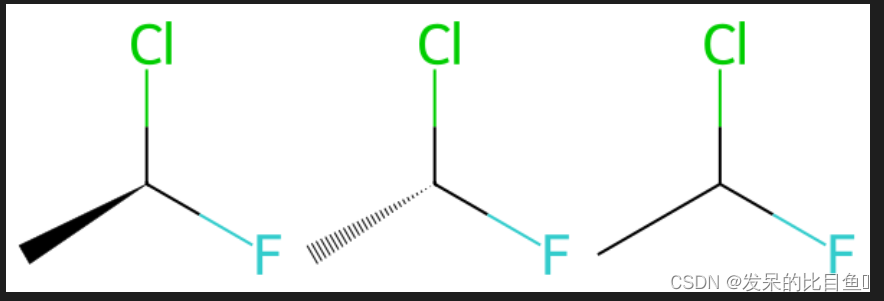
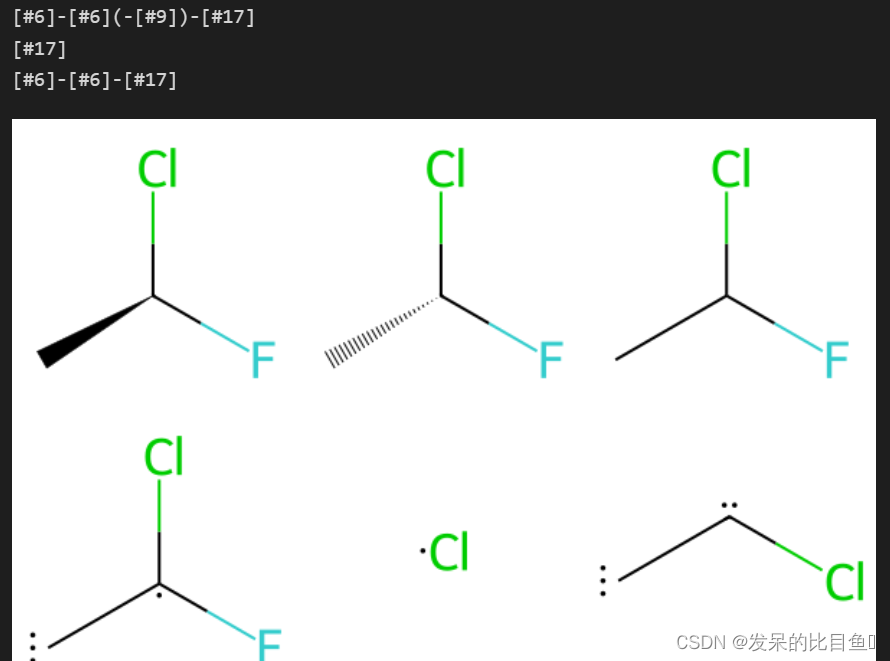
matchChiralTag
# 这是一个具有 的分子@,一个具有 的分子@@,以及一个其中立体未指定的分子。
ms = [Chem.MolFromSmiles(x) for x in ('C[C@H](F)Cl','C[C@@H](F)Cl','CC(F)Cl')]
Draw.MolsToGridImage(ms)
# 默认的 MCS 正是您所期望的 - 每个原子都匹配:
res = rdFMCS.FindMCS(ms)
print(res.smartsString)
results = ms
results = results + [Chem.MolFromSmiles(res.smartsString)]
## 如果我们使用第一个分子和最后一个分子的 MCS matchChiralTag,我们只会得到一个原子(你得到的原子不是先验明显的):
## 这是因为中心 C 原子不再匹配,因为它在第一个分子中指定了手性,而在最后一个分子中指定了未指定的手性。
res = rdFMCS.FindMCS((ms[0],ms[2]),matchChiralTag=True)
print(res.smartsString)
results = results + [Chem.MolFromSmiles(res.smartsString)]
## 另一方面,如果我们对第一个和第二个分子进行 MCS,我们会得到三原子结果:
## 这里中心碳可以匹配,因为它们在两个分子中都具有指定的手性。但我们只能匹配中心碳的两个邻居,因为当我们添加第三个邻居时,实际的手性本身就开始重要。
res = rdFMCS.FindMCS((ms[0],ms[1]),matchChiralTag=True)
print(res.smartsString)
results = results + [Chem.MolFromSmiles(res.smartsString)]
Draw.MolsToGridImage(results)
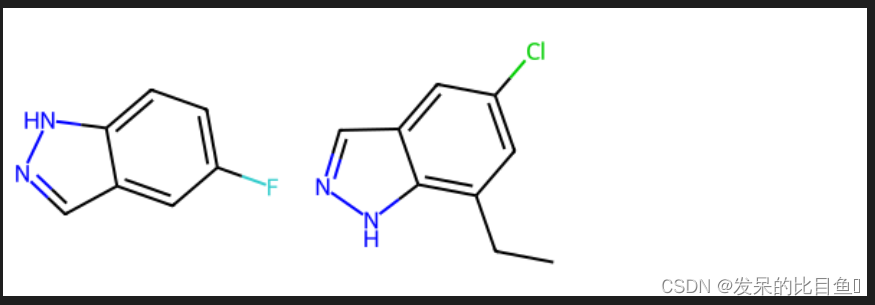
高亮分子的不同子结构
mol1 = Chem.MolFromSmiles('FC1=CC=C2C(=C1)C=NN2')
mol2 = Chem.MolFromSmiles('CCC1=C2NN=CC2=CC(Cl)=C1')
Draw.MolsToGridImage([mol1, mol2])
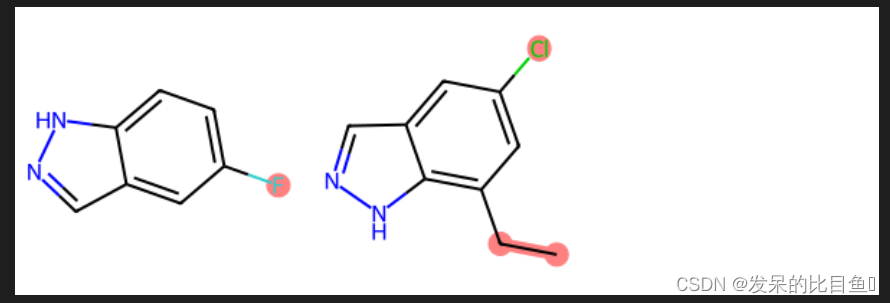
## 定义比较分子不同的方法:
mcs = rdFMCS.FindMCS([mol1,mol2])
mcs_mol = Chem.MolFromSmarts(mcs.smartsString)
match1 = mol1.GetSubstructMatch(mcs_mol)
target_atm1 = []
for atom in mol1.GetAtoms():
if atom.GetIdx() not in match1:
target_atm1.append(atom.GetIdx())
match2 = mol2.GetSubstructMatch(mcs_mol)
target_atm2 = []
for atom in mol2.GetAtoms():
if atom.GetIdx() not in match2:
target_atm2.append(atom.GetIdx())
Draw.MolsToGridImage([mol1, mol2],highlightAtomLists=[target_atm1, target_atm2])
参考
https://rdkit.org/docs/source/rdkit.Chem.rdFMCS.html
https://www.rdkit.org/docs/GettingStartedInPython.html#maximum-common-substructure