文章标题:《Single-cell atlas of lineage states, tumor microenvironment and subtypespecific expression programs in gastric cancer》
DOI: 10.1158/2159-8290.CD-21-0683
数据集组织形式快照:
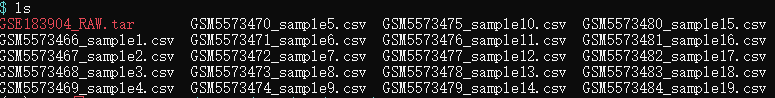
- step1
利用Seurat包整合数据
#! conda env R4
library(Seurat)
library(tidyverse)
library(cowplot)
# output dir
workdir <- ""
# where dataset
projectdir <- ""
file.list <- list.files(projectdir,pattern="csv$")
# create SeuratObject
scelist <-lapply(file.list,function(x){CreateSeuratObject(counts = read.csv(paste(projectdir,x,sep="/"),header=T,row.names=1,sep=","), min.cells = 3,min.features = 200)})
# 把数据集中包含细胞数少于500的去掉
numcol <- lapply(scelist,function(x){ncol(x)})
numcol <- unlist(numcol
scelist <- scelist[numcol > 500]
#log-normalization and identify variable features
sceList1 <- lapply(scelist, function(obj){
obj[["percent.mt"]] <- PercentageFeatureSet(object = obj, pattern = "^MT", assay = 'RNA')
obj <- subset(obj, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 20)
obj <- NormalizeData(obj, normalization.method = "LogNormalize")
obj <- FindVariableFeatures(obj, selection.method = "vst", nfeatures = 2000, verbose = T)
})
features <- SelectIntegrationFeatures(object.list = sceList1)
sceList1 <- lapply(X = sceList1, FUN = function(x) {
x <- ScaleData(x, features = features, verbose = FALSE)
x <- RunPCA(x, features = features,npcs = 30, verbose = FALSE)
})
# then identify anchors using the FindIntegrationAnchors() function, which takes a list of Seurat objects as input
immune.anchors <- FindIntegrationAnchors(object.list = sceList1,reduction = "rpca")
# and use these anchors to integrate the two datasets together with IntegrateData()
immune.combined <- IntegrateData(anchorset = immune.anchors)
immune.combined <- ScaleData(immune.combined, verbose = FALSE)
immune.combined <- RunPCA(immune.combined, npcs = 30, verbose = FALSE)
immune.combined <- RunUMAP(immune.combined, reduction = "pca", dims = 1:30)
immune.combined <- FindNeighbors(immune.combined, reduction = "pca", dims = 1:30)
immune.combined <- FindClusters(immune.combined, resolution = 0.5)
cat("move to SingleR\n")
# singleR 注释
library(celldex)
library(SingleR)
hpca.se <- HumanPrimaryCellAtlasData()
counts <- GetAssayData(immune.combined[["RNA"]], slot="counts")
pred.sce <- SingleR(test = counts, ref = hpca.se, labels = hpca.se$label.main)
immune.combined@meta.data$singleR <- pred.sce$labels
saveRDS(immune.combined,file = paste(workdir,"/Annotation/immune.combined.rds",sep=""))





![NSS [SWPUCTF 2021 新生赛]no_wakeup](https://img-blog.csdnimg.cn/img_convert/0795a515de32c884f6536a9cb0981834.png)



![【数据结构与算法】将含有n个元素的整数数组A[0…n-1]的元素循环右移1≤m<n)位。要求算法的空间复杂度为O(1)。](https://img-blog.csdnimg.cn/0e4a9c7a3f7d4325a3fcbe145698b6d8.png)
![[MySQL]MySQL数据库基础](https://img-blog.csdnimg.cn/img_convert/fae5bf1d4fe0bd9963e7449f831c7aa8.png)