<~生~信~交~流~与~合~作~请~关~注~公~众~号@生信探索>
流程代码在:https://jihulab.com/BioQuest/SnakeMake-mNGS 或https://github.com/BioQuestX/SnakeMake-mNGS
教程链接在:https://doc.bioquest.cn/mngs
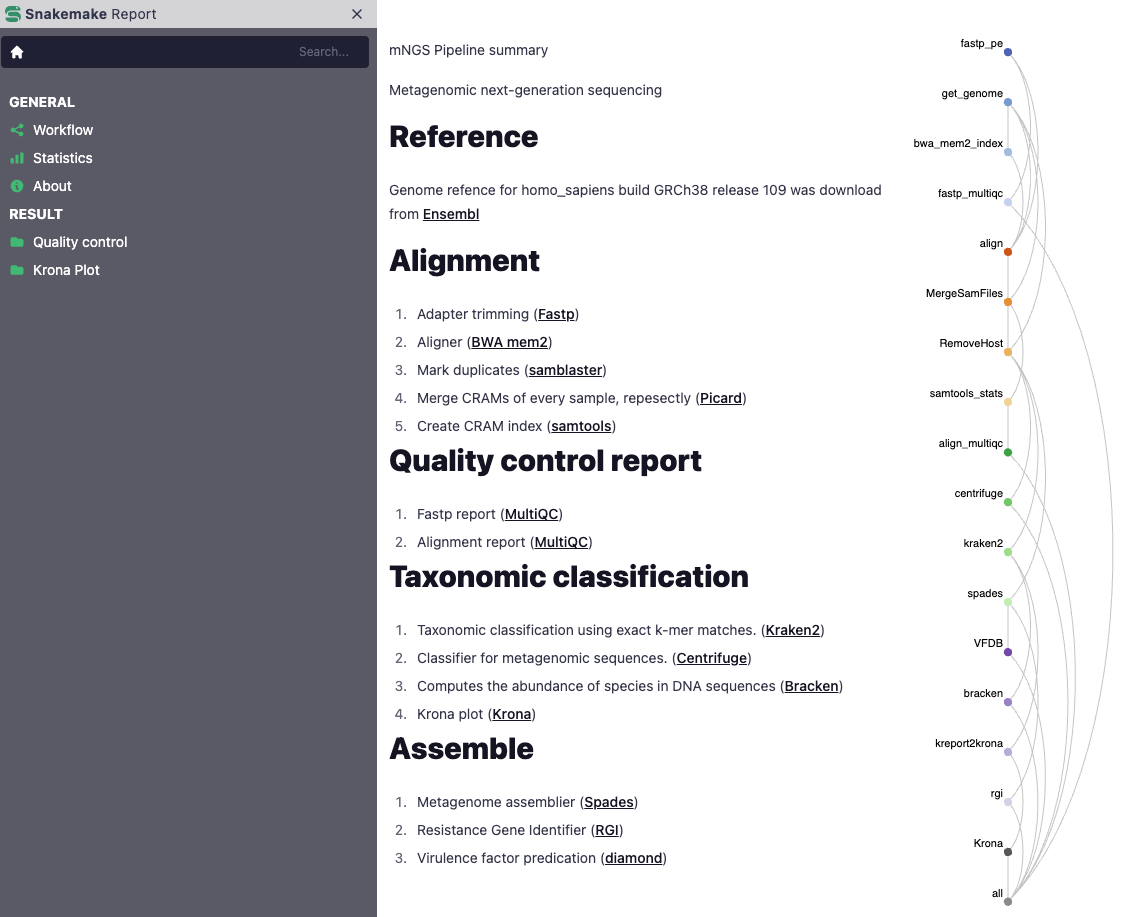
mNGS Pipeline summary
Metagenomic next-generation sequencing
Reference
Genome refence was download from Ensembl
Alignment
-
Adapter trimming ( fastp) -
Aligner ( BWA mem2) -
Mark duplicates ( samblaster) -
Merge CRAMs of every sample, repesectly ( Picard) -
Create CRAM index ( samtools)
Quality control report
-
Fastp report ( MultiQC) -
Alignment report ( MultiQC)
Taxonomic classification
-
Taxonomic classification using exact k-mer matches ( Kraken) -
Classifier for metagenomic sequences ( Centrifuge) -
Computes the abundance of species in DNA sequences ( Bracken) -
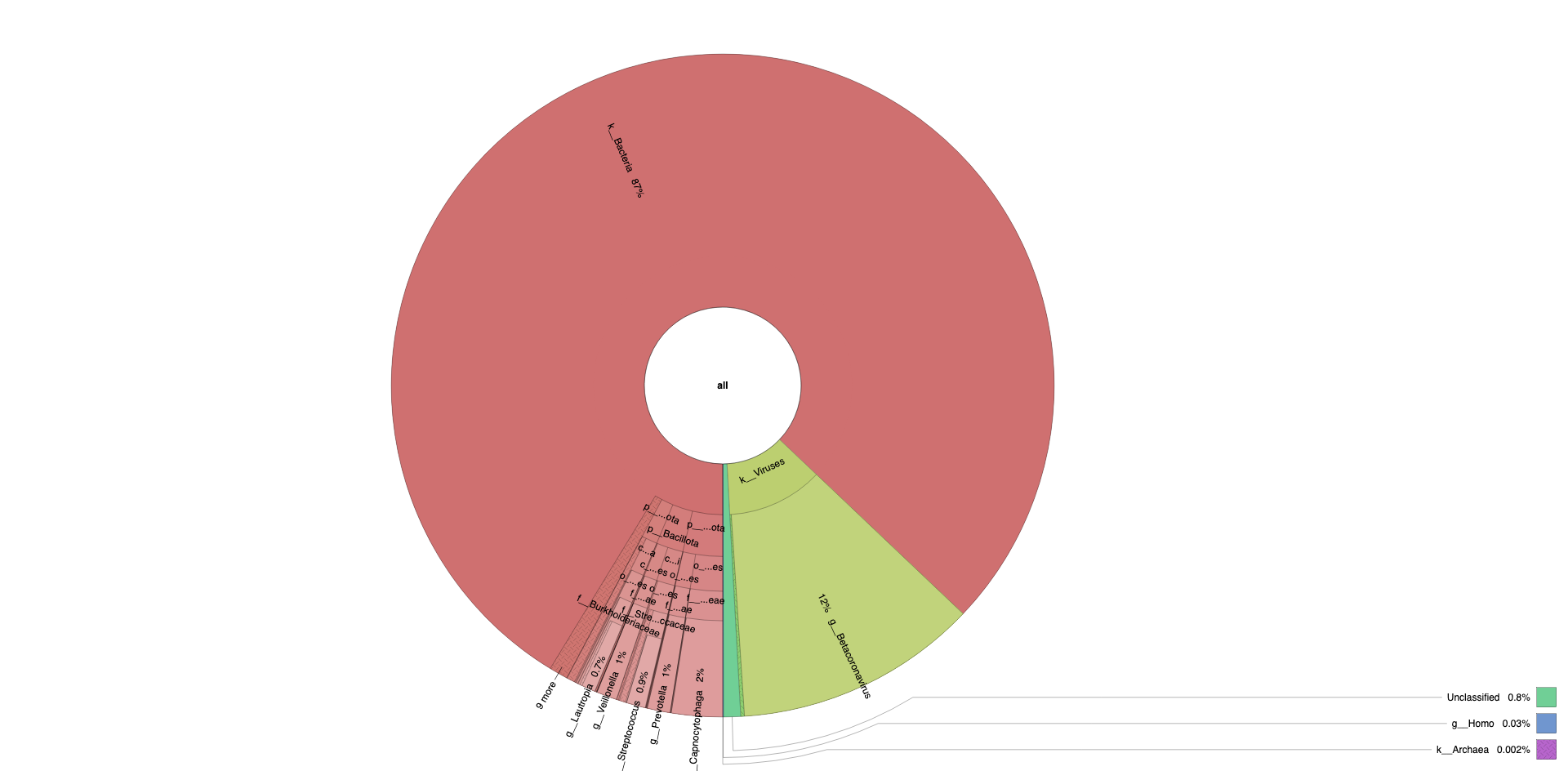
Krona plot ( Krona)
Assemble
-
Metagenome assemblier ( Spades) -
Resistance Gene Identifier ( RGI) -
Virulence factor predication ( diamond)
SnakeMake Report
Outputs
.
├── config
│ ├── config.yaml
│ └── samples.tsv
├── dag.svg
├── logs
│ ├── align
│ ├── bracken
│ ├── centrifuge
│ ├── fastp
│ ├── kraken2
│ ├── kreport2krona
│ ├── Krona
│ ├── MergeSamFiles
│ ├── qc
│ ├── RemoveHost
│ ├── rgi
│ ├── spades
│ └── VFDB
├── raw
│ ├── SRR10903401.1.fastq.gz
│ ├── SRR10903401.2.fastq.gz
│ ├── SRR10903402.1.fastq.gz
│ └── SRR10903402.2.fastq.gz
├── README.md
├── report
│ ├── align_multiqc_data
│ ├── align_multiqc.html
│ ├── bracken
│ ├── centrifuge
│ ├── fastp_multiqc_data
│ ├── fastp_multiqc.html
│ ├── kraken2
│ └── Krona
├── report.html
├── results
│ ├── aligned
│ ├── centrifuge
│ ├── kraken2
│ ├── kreport2krona
│ ├── rgi
│ ├── spades
│ └── VFDB
└── workflow
├── envs
├── report
├── rules
├── schemas
├── scripts
└── Snakefile
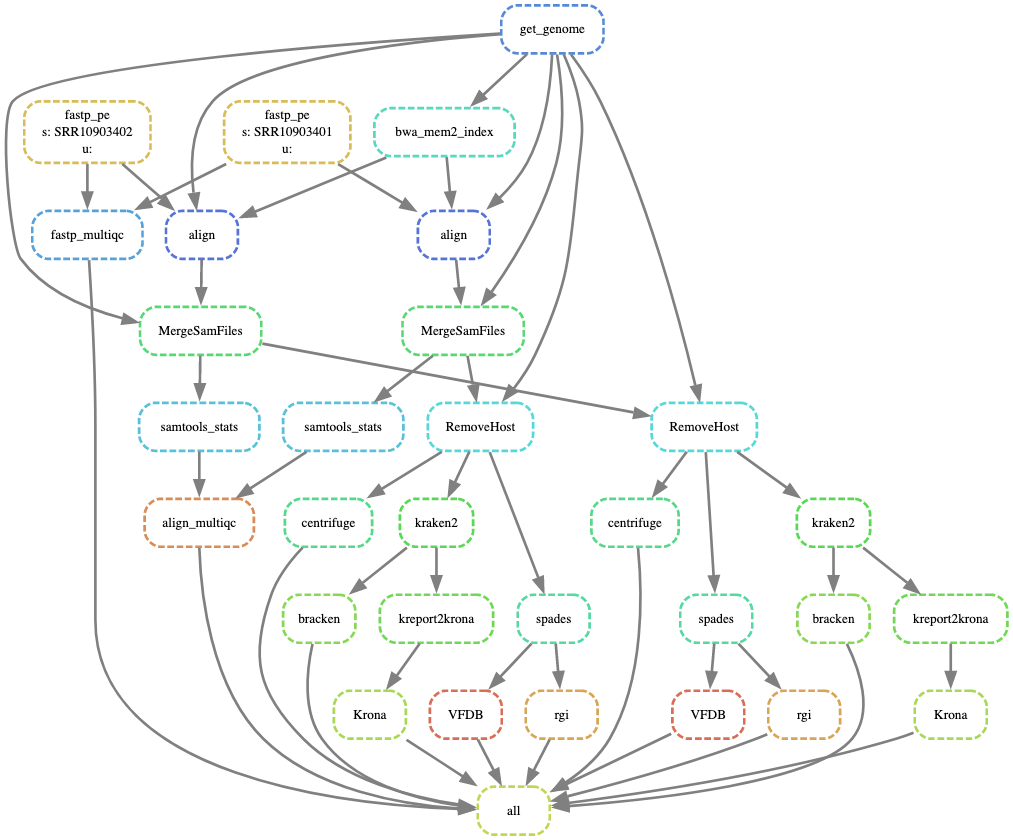
Directed Acyclic Graph
Krona Plot
本文由 mdnice 多平台发布