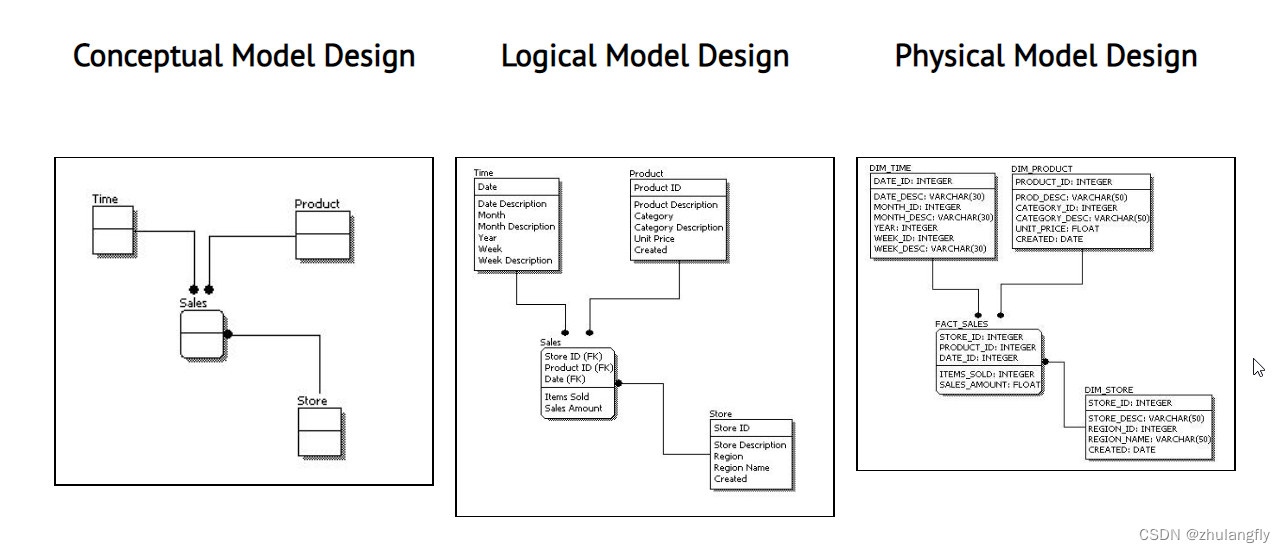
1 生物信息学简介
生物信息学(BioInformatics)是研究生物信息的采集、处理、存储、传播,分析和解释等各方面的学科,也是随着生命科学和计算机科学的迅猛发展,生命科学和计算机科学相结合形成的一门新学科。它通过综合利用生物学,计算机科学和信息技术而揭示大量而复杂的生物数据所赋有的生物学奥秘。
生物信息学是建立在分子生物学的基础上的,因此,要了解生物信息学,就必须先对分子生物学的发展有一个简单的了解。研究生物细胞的生物大分子的结构与功能很早就已经开始,1866年孟德尔从实验上提出了假设:遗传因子是以生物成分存在,1871年Miescher从死的白细胞核中分离出脱氧核糖核酸(DNA),在Avery和McCarty于1944年证明了DNA是生命器官的遗传物质以前,人们仍然认为染色体蛋白质携带基因,而DNA是一个次要的角色。1944年Chargaff发现了著名的Chargaff规律,即DNA中鸟嘌呤的量与胞嘧定的量总是相等,腺嘌呤与胸腺嘧啶的量相等。与此同时,Wilkins与Franklin用X射线衍射技术测定了DNA纤维的结构。1953年James Watson和Francis Crick在Nature杂志上推测出DNA的三维结构(双螺旋)。DNA以磷酸糖链形成发双股螺旋,脱氧核糖上的碱基按Chargaff规律构成双股磷酸糖链之间的碱基对。这个模型表明DNA具有自身互补的结构,根据碱基对原则,DNA中贮存的遗传信息可以精确地进行复制。他们的理论奠定了分子生物学的基础。DNA双螺旋模型已经预示出了DNA复制的规则,Kornberg于1956年从大肠杆菌(E.coli)中分离出DNA聚合酶I(DNA polymerase I),能使4种dNTP连接成DNA。DNA的复制需要一个DNA作为模板。Meselson与Stahl(1958)用实验方法证明了DNA复制是一种半保留复制。Crick于1954年提出了遗传信息传递的规律,DNA是合成RNA的模板,RNA又是合成蛋白质的模板,称之为中心法则(Central dogma),这一中心法则对以后分子生物学和生物信息学的发展都起到了极其重要的指导作用。经过Nirenberg和Matthai(1963)的努力研究,编码20氨基酸的遗传密码得到了破译。限制性内切酶的发现和重组DNA的克隆(clone)奠定了基因工程的技术基础。正是由于分子生物学的研究对生命科学的发展有巨大的推动作用,生物信息学的出现也就成了一种必然。2001年2月,人类基因组工程测序的完成,使生物信息学走向了一个高潮。由于DNA自动测序技术的快速发展,DNA数据库中的核酸序列公共数据量以每天106bp速度增长,生物信息迅速地膨胀成数据的海洋。毫无疑问,我们正从一个积累数据向解释数据的时代转变,数据量的巨大积累往往蕴含着潜在突破性发现的可能,“生物信息学”正是从这一前提产生的交叉学科。粗略地说,该领域的核心内容是研究如何通过对DNA序列的统计计算分析,更加深入地理解DNA序列,结构,演化及其与生物功能之间的关系,其研究课题涉及到分子生物学,分子演化及结构生物学,统计学及计算机科学等许多领域。生物信息学是内涵非常丰富的学科,其核心是基因组信息学,包括基因组信息的获取,处理,存储,分配和解释。基因组信息学的关键是“读懂”基因组的核苷酸顺序,即全部基因在染色体上的确切位置以及各DNA片段的功能;同时在发现了新基因信息之后进行蛋白质空间结构模拟和预测,然后依据特定蛋白质的功能进行药物设计。了解基因表达的调控机理也是生物信息学的重要内容,根据生物分子在基因调控中的作用,描述人类疾病的诊断,治疗内在规律。它的研究目标是揭示“基因组信息结构的复杂性及遗传语言的根本规律”,解释生命的遗传语言。生物信息学已成为整个生命科学发展的重要组成部分,成为生命科学研究的前沿。
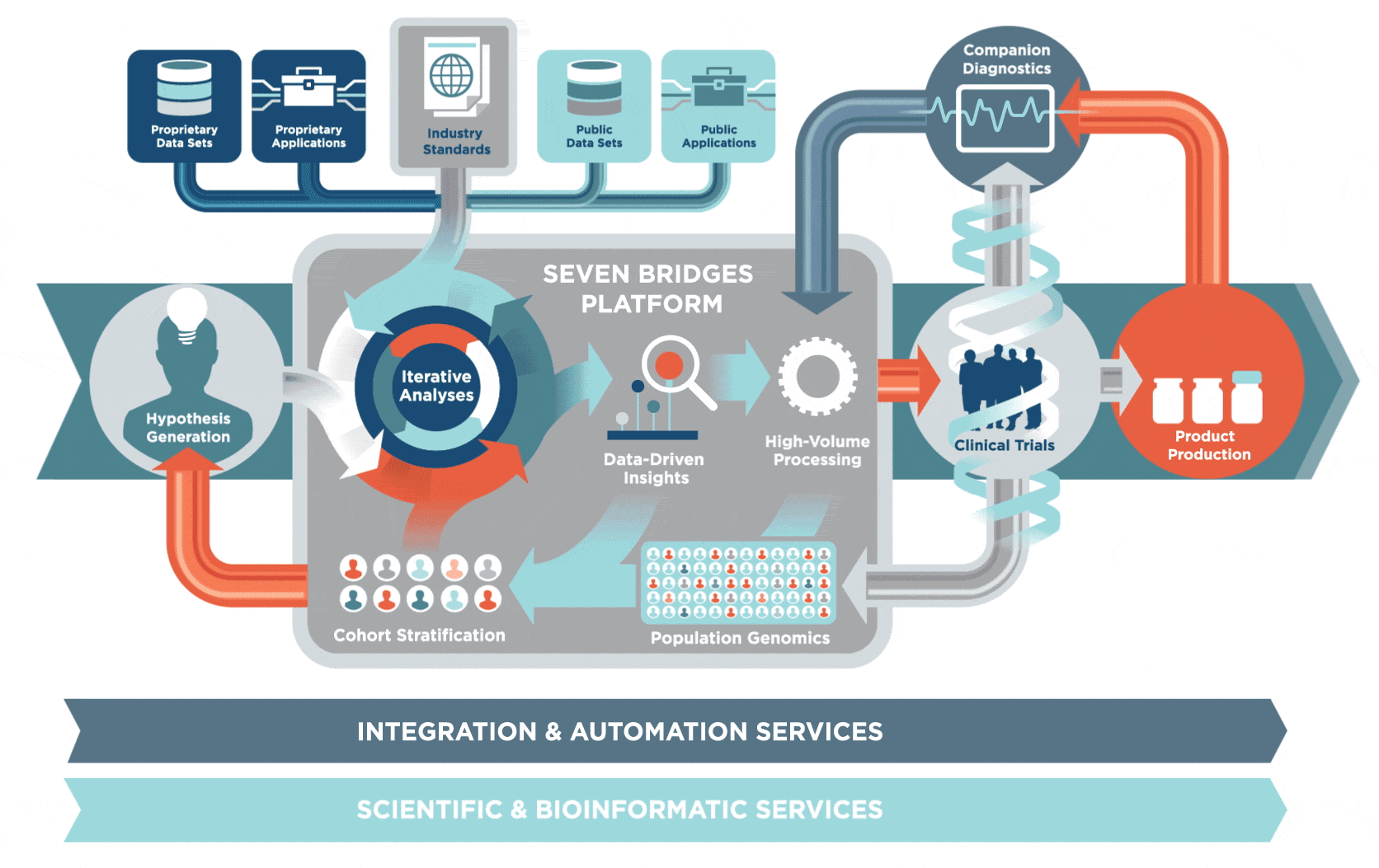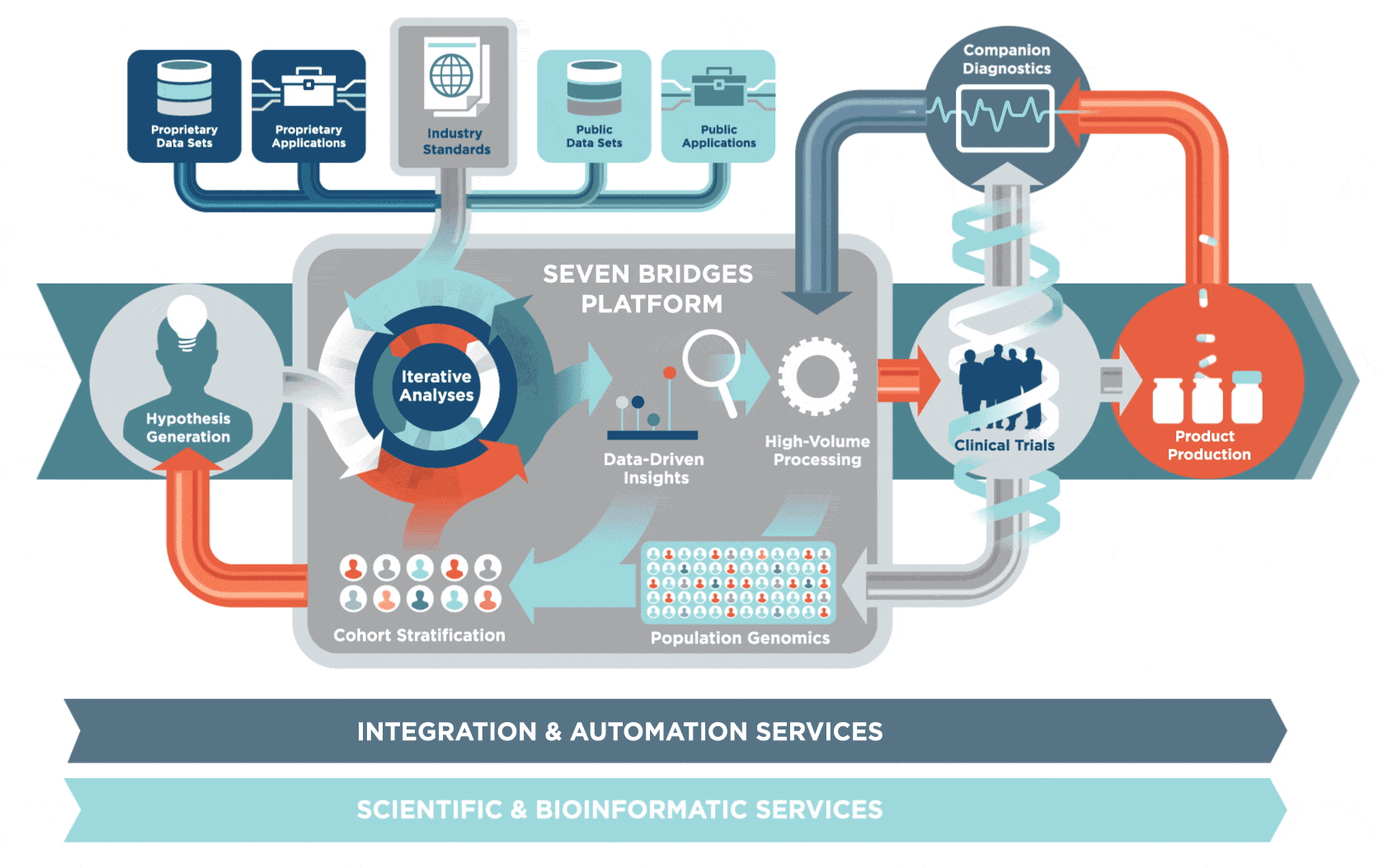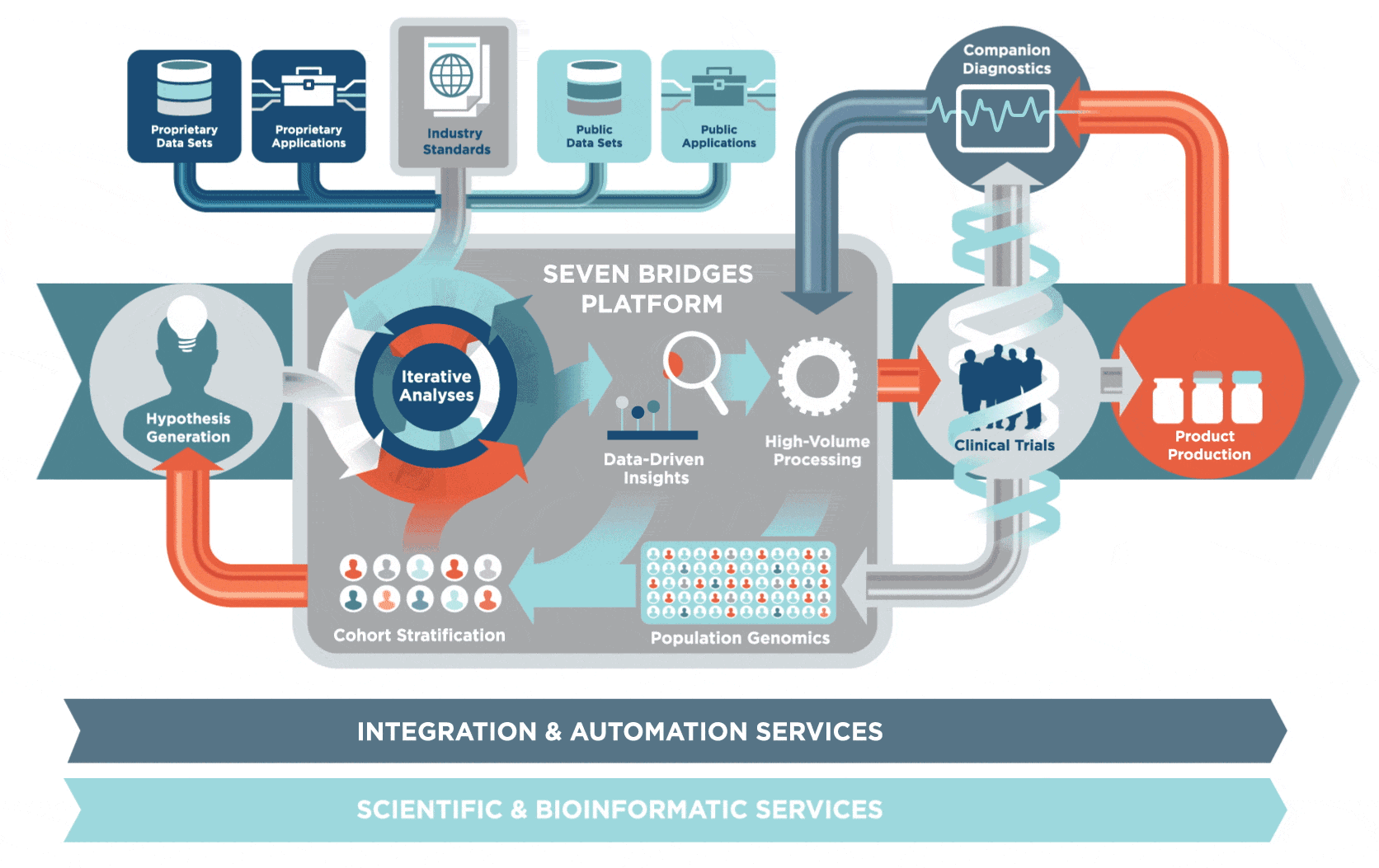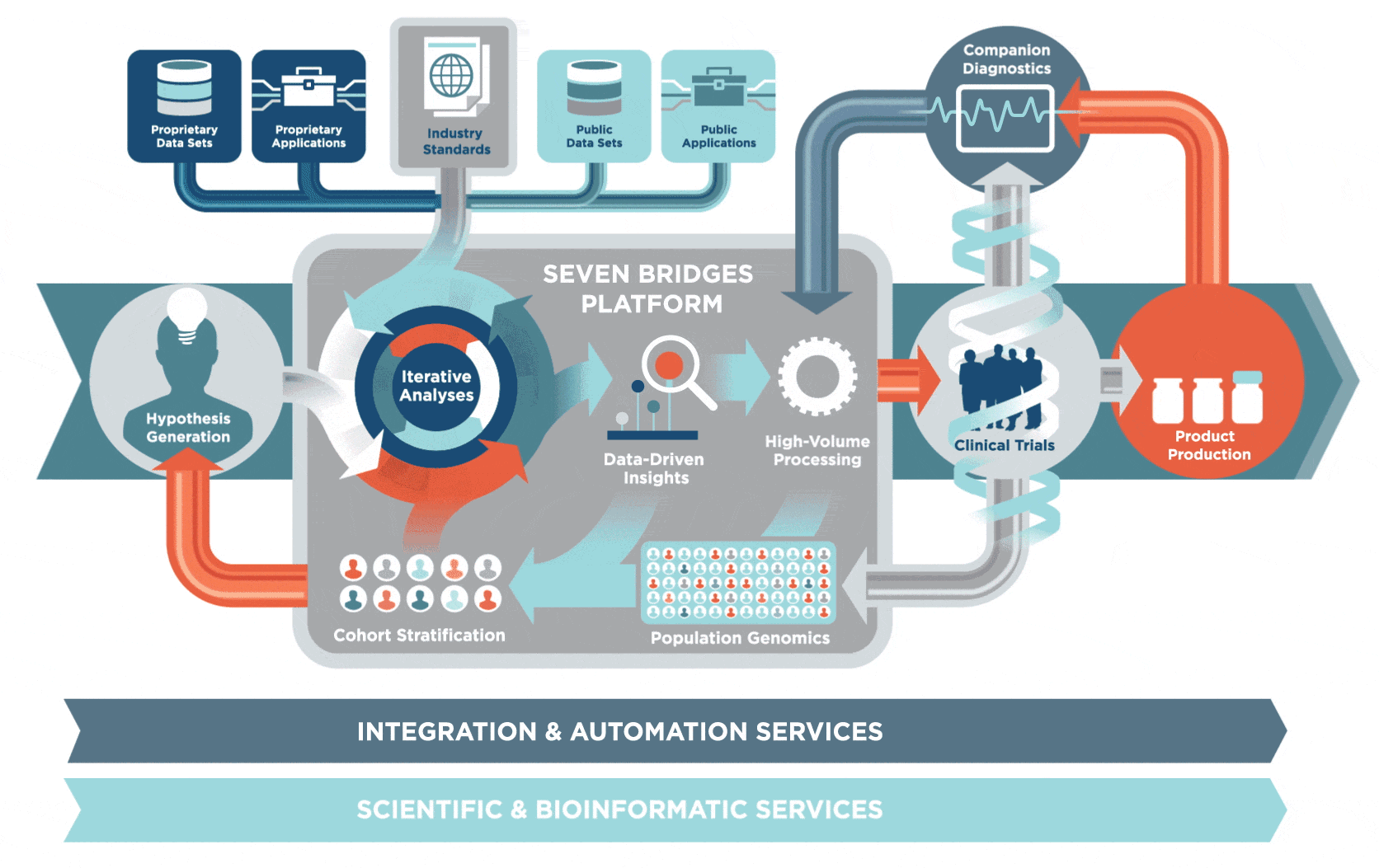
2 生物信息学研究方向
生物信息学在短短十几年间,已经形成了多个研究方向,以下简要介绍一些主要的研究重点。
2.1 序列比对
序列比对(Sequence Alignment)的基本问题是比较两个或两个以上符号序列的相似性或不相似性。从生物学的初衷来看,这一问题包含了以下几个意义:从相互重叠的序列片断中重构DNA的完整序列。在各种试验条件下从探测数据(probe data)中决定物理和基因图存贮,遍历和比较数据库中的DNA序列,比较两个或多个序列的相似性,在数据库中搜索相关序列和子序列,寻找核苷酸(nucleotides)的连续产生模式,找出蛋白质和DNA序列中的信息成分。序列比对考虑了DNA序列的生物学特性,如序列局部发生的插入,删除(前两种简称为indel)和替代,序列的目标函数获得序列之间突变集最小距离加权和或最大相似性和,对齐的方法包括全局对齐,局部对齐,代沟惩罚等。两个序列比对常采用动态规划算法,这种算法在序列长度较小时适用,然而对于海量基因序列(如人的DNA序列高达10^9bp),这一方法就不太适用,甚至采用算法复杂性为线性的也难以奏效。因此,启发式方法的引入势在必然,著名的BLAST和FASTA算法及相应的改进方法均是从此前提出发的。
2.2 蛋白质比对
基本问题是比较两个或两个以上蛋白质分子空间结构的相似性或不相似性。蛋白质的结构与功能是密切相关的,一般认为,具有相似功能的蛋白质结构一般相似。蛋白质是由氨基酸组成的长链,长度从50到1000~3000AA(Amino Acids),蛋白质具有多种功能,如酶,物质的存贮和运输,信号传递,抗体等等。氨基酸的序列内在的决定了蛋白质的3维结构。一般认为,蛋白质有四级不同的结构。研究蛋白质结构和预测的理由是:医药上可以理解生物的功能,寻找dockingdrugs的目标,农业上获得更好的农作物的基因工程,工业上有利用酶的合成。直接对蛋白质结构进行比对的原因是由于蛋白质的3维结构比其一级结构在进化中更稳定的保留,同时也包含了较AA序列更多的信息。蛋白质3维结构研究的前提假设是内在的氨基酸序列与3维结构一一对应(不一定全真),物理上可用最小能量来解释。从观察和总结已知结构的蛋白质结构规律出发来预测未知蛋白质的结构。同源建模(homology modeling)和指认(Threading)方法属于这一范畴。同源建模用于寻找具有高度相似性的蛋白质结构(超过30%氨基酸相同),后者则用于比较进化族中不同的蛋白质结构。然而,蛋白结构预测研究现状还远远不能满足实际需要。
2.3 基因识别分析
基因识别的基本问题是给定基因组序列后,正确识别基因的范围和在基因组序列中的精确位置。非编码区由内含子组成(introns),一般在形成蛋白质后被丢弃,但从实验中,如果去除非编码区,又不能完成基因的复制。显然,DNA序列作为一种遗传语言,既包含在编码区,又隐含在非编码序列中。分析非编码区DNA序列没有一般性的指导方法。在人类基因组中,并非所有的序列均被编码,即是某种蛋白质的模板,已完成编码部分仅占人类基因总序列的3~5%,显然,手工的搜索如此大的基因序列是难以想象的。侦测密码区的方法包括测量密码区密码子(codon)的频率,一阶和二阶马尔可夫链,ORF(Open Reading Frames),启动子(promoter)识别,HMM(Hidden Markov Model)和GENSCAN,Splice Alignment等等。
2.4 分子进化
分子进化是利用不同物种中同一基因序列的异同来研究生物的进化,构建进化树。既可以用DNA序列也可以用其编码的氨基酸序列来做,甚至于可通过相关蛋白质的结构比对来研究分子进化,其前提假定是相似种族在基因上具有相似性。通过比较可以在基因组层面上发现哪些是不同种族中共同的,哪些是不同的。早期研究方法常采用外在的因素,如大小,肤色,肢体的数量等等作为进化的依据。较多模式生物基因组测序任务的完成,人们可从整个基因组的角度来研究分子进化。在匹配不同种族的基因时,一般须处理三种情况:Orthologous:不同种族,相同功能的基因;Paralogous:相同种族,不同功能的基因;Xenologs:有机体间采用其他方式传递的基因,如被病毒注入的基因。这一领域常采用的方法是构造进化树,通过基于特征(即DNA序列或蛋白质中的氨基酸的碱基的特定位置)和基于距离(对齐的分数)的方法和一些传统的聚类方法(如UPGMA)来实现。
2.5 序列重叠群(Contigs)装配
根据现行的测序技术,每次反应只能测出500或更多一些碱基对的序列,如人类基因的测量就采用了短枪(shortgun)方法,这就要求把大量的较短的序列全体构成了重叠群(Contigs)。逐步把它们拼接起来形成序列更长的重叠群,直至得到完整序列的过程称为重叠群装配。从算法层次来看,序列的重叠群是一个NP-完全问题。
2.6 遗传密码
通常对遗传密码的研究认为,密码子与氨基酸之间的关系是生物进化历史上一次偶然的事件而造成的,并被固定在现代生物的共同祖先里,一直延续至今。不同于这种“冻结”理论,有人曾分别提出过选择优化,化学和历史等三种学说来解释遗传密码。随着各种生物基因组测序任务的完成,为研究遗传密码的起源和检验上述理论的真伪提供了新的素材。
2.7 药物设计
人类基因工程的目的之一是要了解人体内约10万种蛋白质的结构,功能,相互作用以及与各种人类疾病之间的关系,寻求各种治疗和预防方法,包括药物治疗。基于生物大分子结构及小分子结构的药物设计是生物信息学中的极为重要的研究领域。为了抑制某些酶或蛋白质的活性,在已知其蛋白质3级结构的基础上,可以利用分子对齐算法,在计算机上设计抑制剂分子,作为候选药物。这一领域目的是发现新的基因药物,有着巨大的经济效益。
2.8 生物系统
随着大规模实验技术的发展和数据累积,从全局和系统水平研究和分析生物学系统,揭示其发展规律已经成为后基因组时代的另外一个研究热点-系统生物学。目前来看,其研究内容包括生物系统的模拟(Curr Opin Rheumatol,2007,463-70),系统稳定性分析(Nonlinear Dynamics Psychol Life Sci,2007,413-33),系统鲁棒性分析(Ernst Schering Res Found Workshop, 2007,69-88)等方面。以SBML(Bioinformatics,2007,1297-8)为代表的建模语言在迅速发展之中,以布尔网络 (PLoS Comput Biol,2007,e163)、微分方程(Mol Biol Cell,2004,3841-62)、随机过程(Neural Comput,2007,3262-92)、离散动态事件系统等(Bioinformatics,2007,336-43)方法在系统分析中已经得到应用。很多模型的建立借鉴了电路和其它物理系统建模的方法,很多研究试图从信息流、熵和能量流等宏观分析思想来解决系统的复杂性问题(Anal Quant Cytol Histol,2007,296-308)。当然,建立生物系统的理论模型还需要很长时间的努力,实验观测数据虽然在海量增加,但是生物系统的模型辨识所需要的数据远远超过了数据的产出能力。例如,对于时间序列的芯片数据,采样点的数量还不足以使用传统的时间序列建模方法,巨大的实验代价是系统建模主要困难。系统描述和建模方法也需要开创性的发展。
2.9 技术方法
生物信息学不仅仅是生物学知识的简单整理和数学、物理学、信息科学等学科知识的简单应用。海量数据和复杂的背景导致机器学习、统计数据分析和系统描述等方法需要在生物信息学所面临的背景之中迅速发展。巨大的计算量、复杂的噪声模式、海量的时变数据给传统的统计分析带来了巨大的困难,需要像非参数统计(BMC Bioinformatics,2007,339)、聚类分析(Qual Life Res,2007,1655-63)等更加灵活的数据分析技术。高维数据的分析需要偏最小二乘(partial least squares,PLS)等特征空间的压缩技术。在计算机算法的开发中,需要充分考虑算法的时间和空间复杂度,使用并行计算、网格计算等技术来拓展算法的可实现性。
2.10 其他
如基因表达谱分析,代谢网络分析;基因芯片设计和蛋白质组学数据分析等,逐渐成为生物信息学中新兴的重要研究领域;在学科方面,由生物信息学衍生的学科包括结构基因组学,功能基因组学,比较基因组学,蛋白质学,药物基因组学,中药基因组学,肿瘤基因组学,分子流行病学和环境基因组学,成为系统生物学的重要研究方法。从发展不难看出,基因工程已经进入了后基因组时代。我们也有应对与生物信息学密切相关的如机器学习,和数学中可能存在的误导有一个清楚的认识。
3 序列文件(如FASTA)合并工具开发实践
文件合并,比如fasta文件的合并,经常有于进行密码子使用频率的计算。
另外,合并Dataframe在生信分析项目开发中也经常会用到,以合并多个样本的的两种.tsv文件结果为例,演示如何进行tsv文件内容的读取,并且合并为一个Dataframe,然后保存。
分别比对每个 read group 然后将得到的比对文件合并为一个等等。
3.1 文件合并工具功能需求
1、合并指定的一系列文件,成为一个大文件;
2、或 合并指定文件夹下的所有文件,成为一个大文件;这种方式更好;
3、能合并大量的文件,比如1000个 或更多;
4、能看到运行的进度,不能只看到等待鼠标状态;
5、尽可能自动化,比如自动后缀,自动合并文件名等等;
6、软件要稳定可靠;
3.2 核心代码
using System;
using System.IO;
using System.Text;
using System.Linq;
using System.Drawing;
using System.Collections;
using System.Collections.Generic;
namespace Legal.BIOG
{
public static partial class Tools
{
/// <summary>
/// 合并文件(支持超多、超大文件)
/// </summary>
/// <param name="sourceFiles">要合并的文件集合</param>
/// <param name="combinedFIle">合并后的文件名称</param>
private static void CombineFiles(string[] sourceFiles, string combinedFIle)
{
FileStream src = new FileStream(combinedFIle, FileMode.Append);
BinaryWriter sw = new BinaryWriter(src);
for (int i = 0; i < sourceFiles.Length; i++)
{
FileStream dst = new FileStream(sourceFiles[i].ToString(), FileMode.Open);
BinaryReader dr = new BinaryReader(dst);
sw.Write(dr.ReadBytes((int)dst.Length));
dr.Close();
dst.Close();
}
sw.Close();
src.Close();
}
}
}
3.3 运行效果
文件合并工具是北京联高软件开发有限公司出品的生信初级软件 BIOG 的功能之一。
BIOG 基本功能:

文件合并实例:
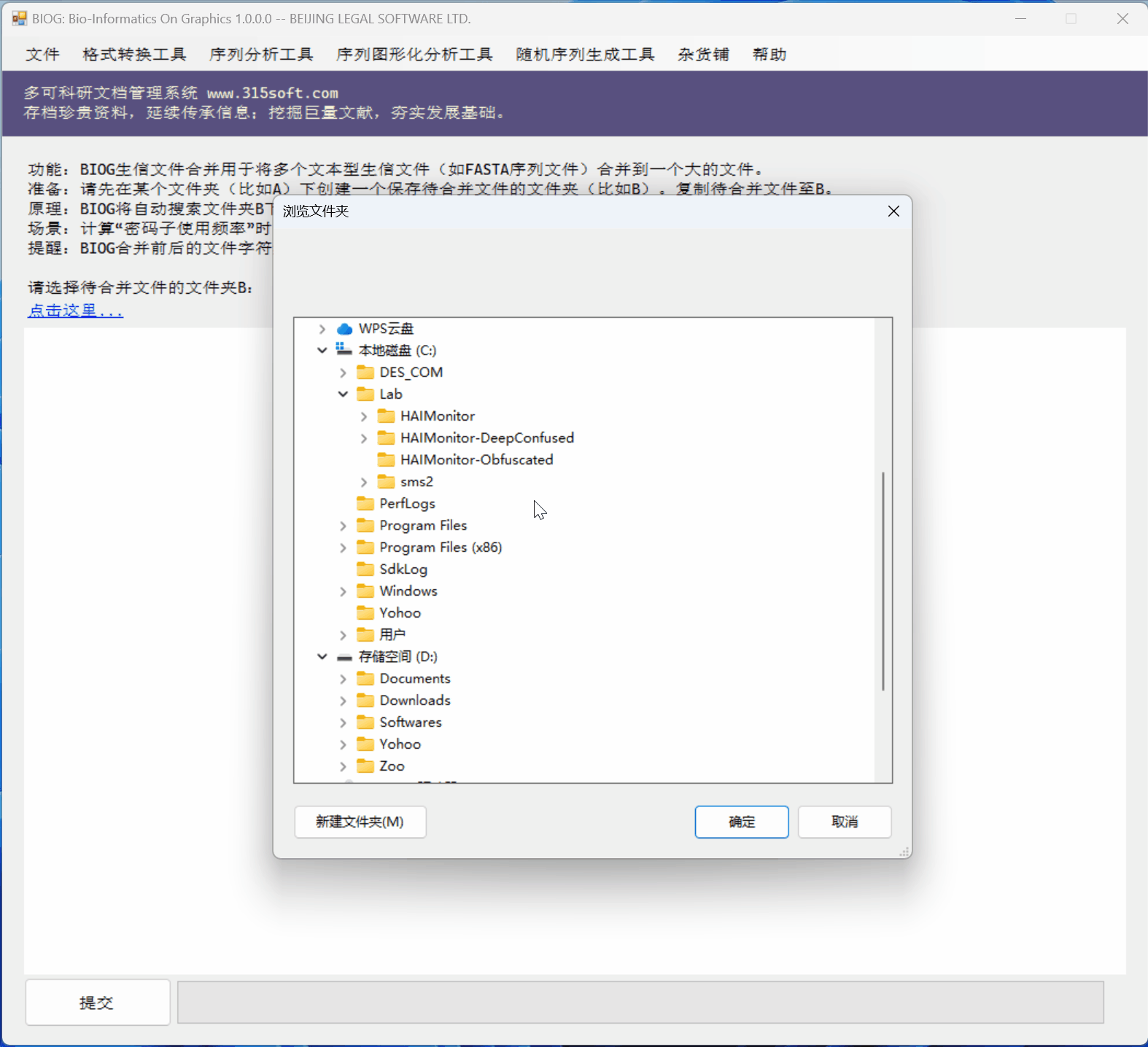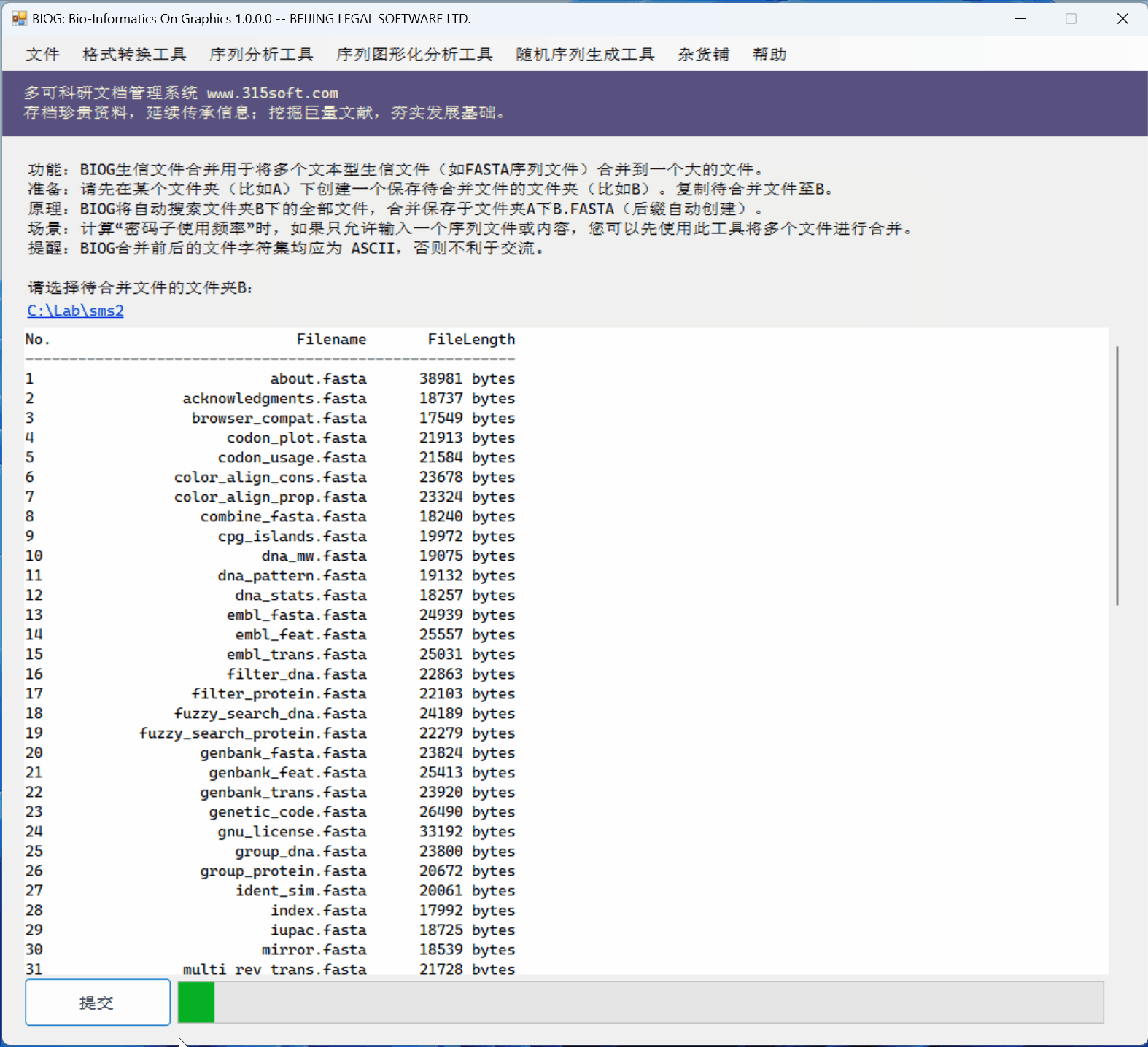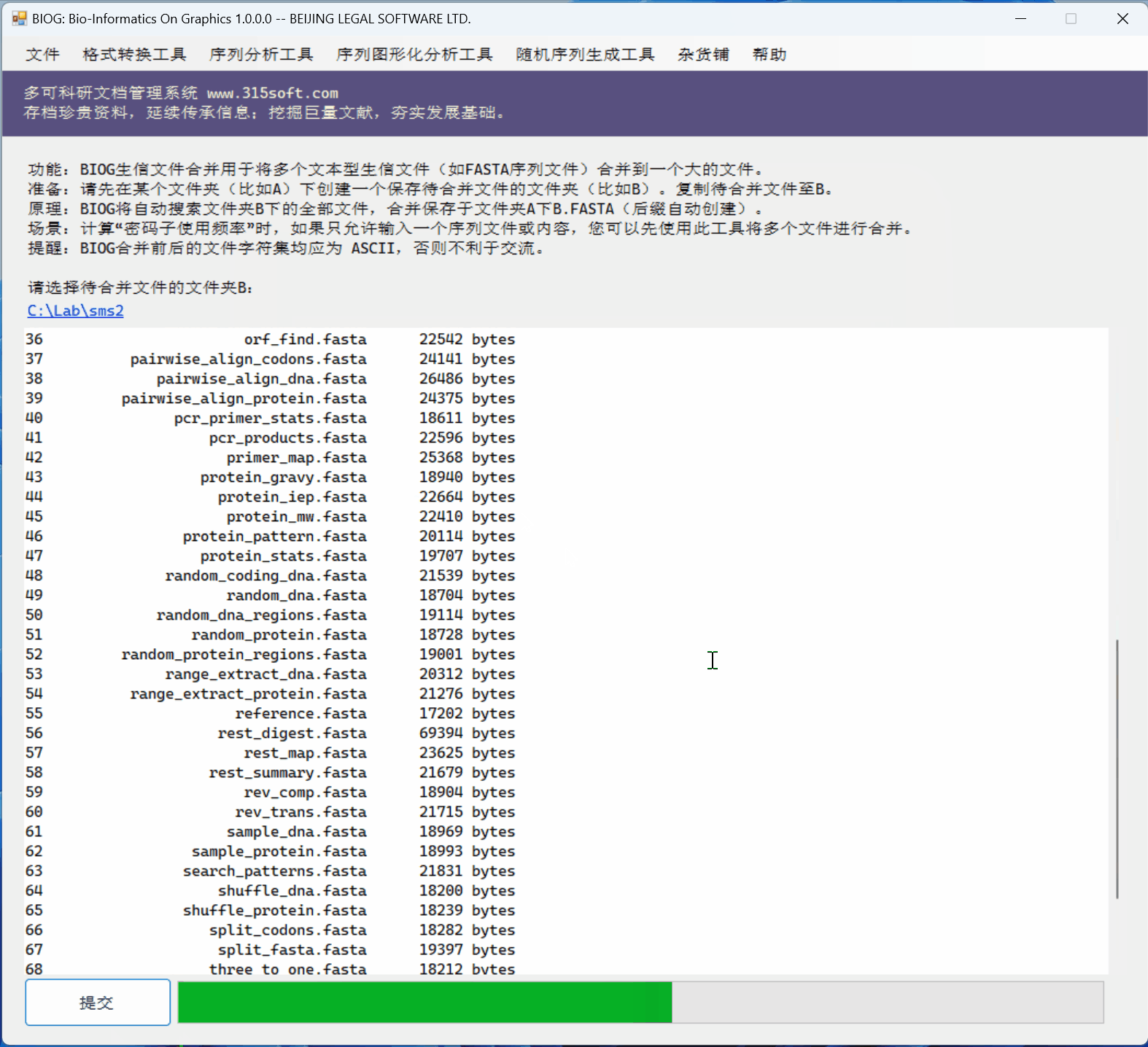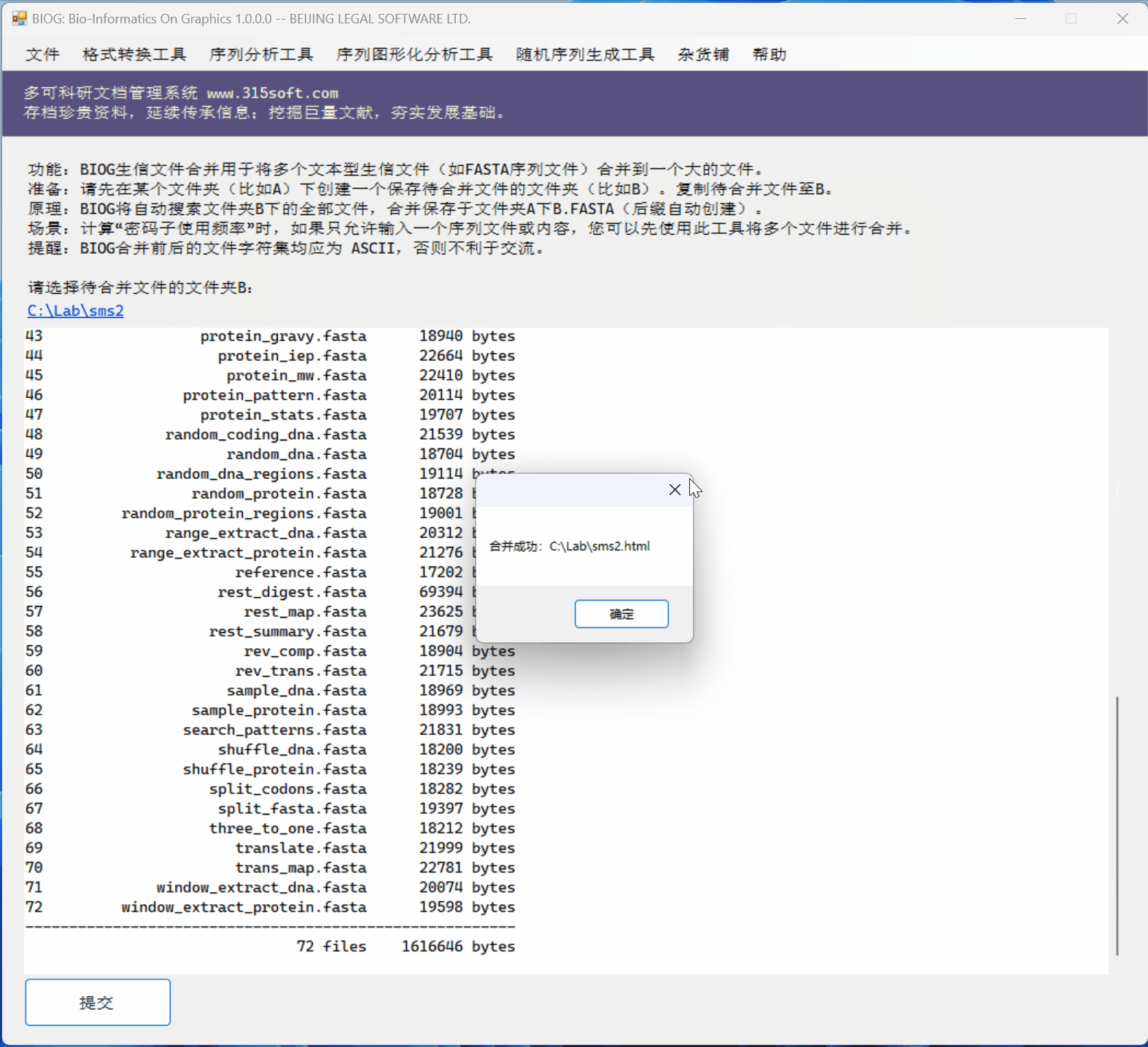
(上面图片为模拟数据,慢速运行,实际速度快很多)
3.4 桌面软件与在线软件的一些区别
不少 生信软件(工具型,规模小的)以在线模式提供,其优点是:
(1)无需安装,直接使用;
(2)永远是最新版本;
(3)问题与反馈及时;
但也存在明显的缺陷:
(1)无法处理大量的或大的数据;
(2)处理进程无法获得及时响应,往往不知道处理到哪个进度了;
(3)有很多不便利之处;
手边有一个桌面型生信软件作为辅助工具,不失为一个选择。