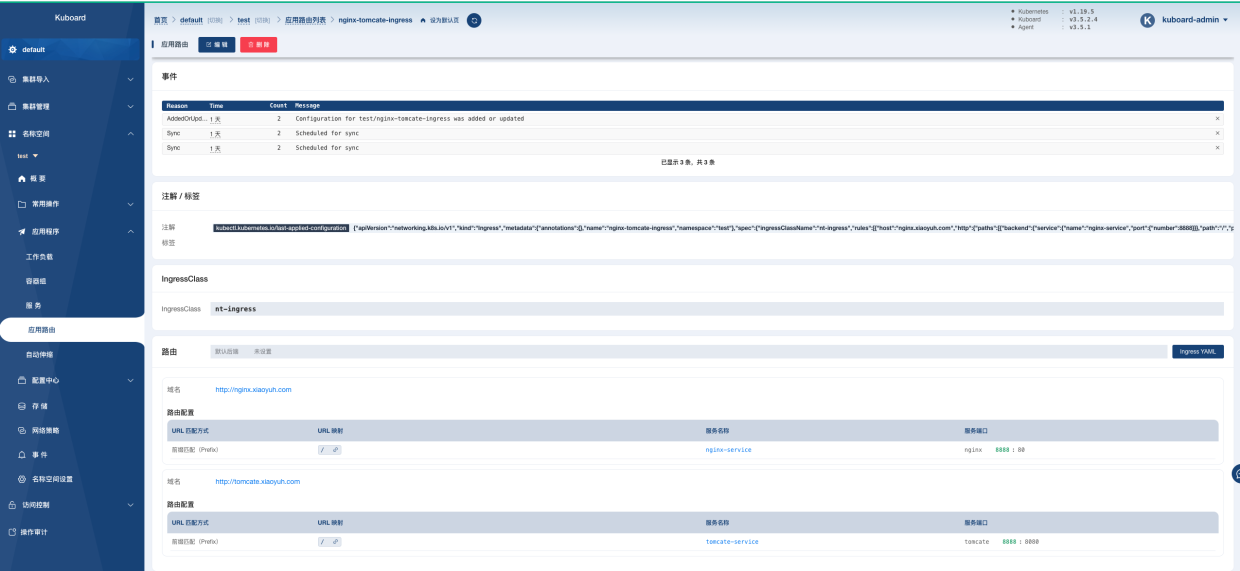
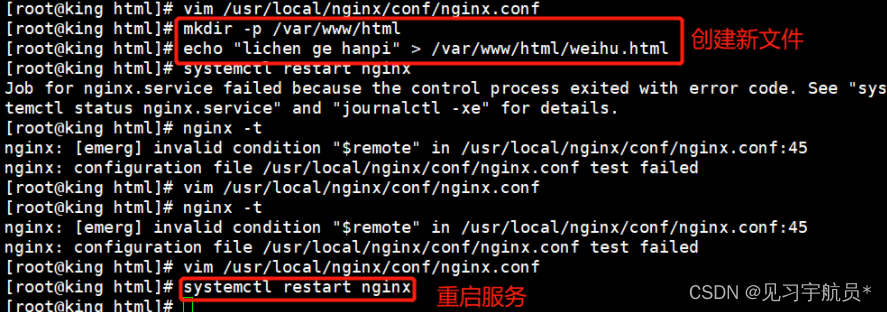
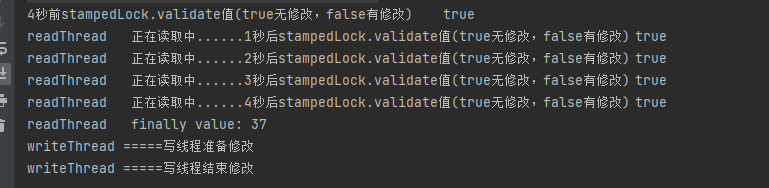
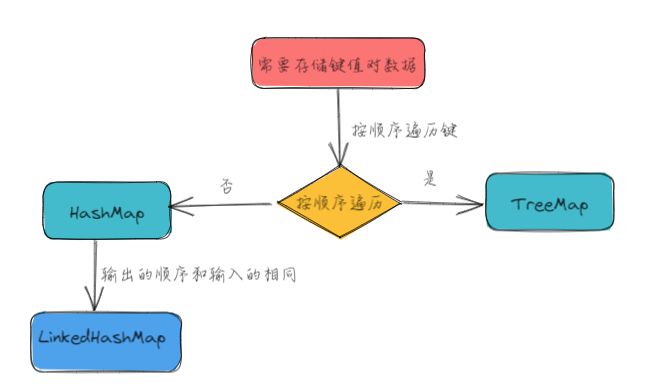
第一性原理差分电荷密度(DFT)计算是一种用于计算分子和材料结构的计算化学方法。它基于物理学原理和量子力学理论,通过解决薛定谔方程来计算电子在分子和材料结构中的行为。DFT是一种非常重要的计算方法,因为它可以提供关于分子和材料结构的详细信息,这些信息可以用于研究化学反应、物质性质和许多其他方面的研究。
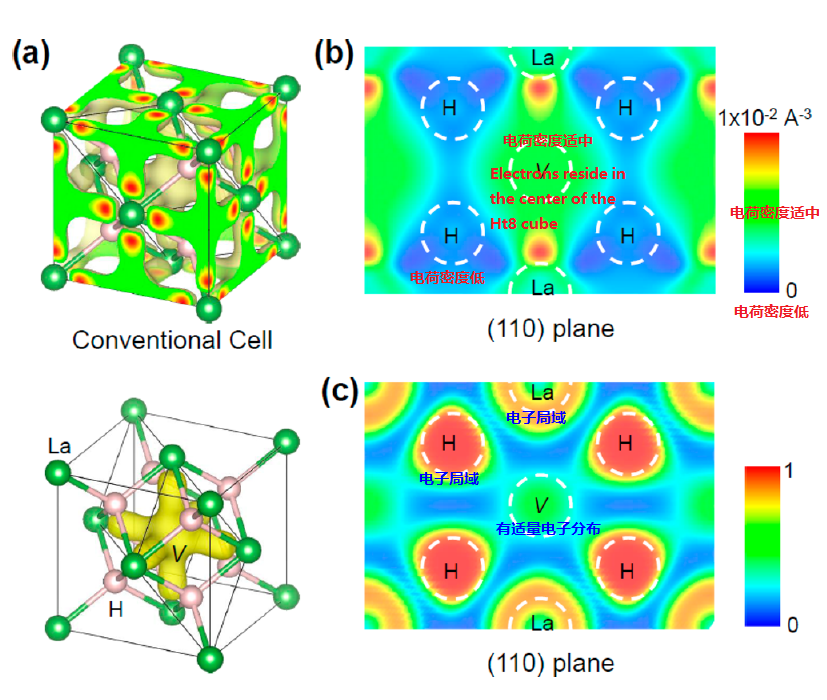
DFT计算通过计算分子或材料结构中的每个原子的电子态来实现。这些电子态可以用电荷密度函数表示。电荷密度函数是描述电子在分子或材料结构中的分布的数学函数。
通过计算电荷密度函数,DFT可以提供一系列有用的信息,如:
能带结构:DFT计算可以提供分子或材料结构中的电子能带结构,即电子能量随动量的变化曲线。这些能带结构可以提供有关分子或材料结构的电子性质的重要信息。
电子密度分布:DFT计算可以提供分子或材料结构中的电子密度分布。这些信息可以用于确定分子或材料结构的电子云形状和大小。
光谱性质:DFT计算可以提供关于分子或材料结构的各种光谱性质的信息,如吸收光谱和紫外光谱。这些信息可以用于确定分子或材料结构的光学性质。
化学反应:DFT计算可以用于研究化学反应的动力学和热力学性质。这些信息可以用于设计更有效的化学反应或更有效的化学反应催化剂。
分子或材料性质:DFT计算可以提供分子或材料结构的各种性质的信息,如电荷分布、电荷密度、分子形状、分子重量和化学键能量。这些信息可以用于确定分子或材料结构的性质,从而为设计更有效的分子或材料提供有用的信息。
在进行DFT计算时,需要使用一些软件工具。最常用的软件工具之一是VASP(Vienna Ab initio Simulation Package)。VASP可以用于计算分子和材料结构的电子性质,并提供各种信息,如电子密度、能带结构、光谱性质和化学反应。其他常用的DFT软件包还包括Gaussian、Quantum Espresso、CRYSTAL、NWChem等等。
除了软件工具,进行DFT计算还需要确定一些参数,例如:
交换-相关泛函:在DFT计算中,需要选择适当的交换-相关泛函来描述电子的交换和相关作用。常用的交换-相关泛函包括局域密度近似(LDA)、广义梯度近似(GGA)和杂化泛函等。
基组:在DFT计算中,需要使用基组来描述原子轨道和电子密度分布。常用的基组包括赝势基组和真实基组。
网格:在DFT计算中,需要将电荷密度函数在空间中离散化为一系列网格点。网格的密度越高,计算结果的精度越高,但计算所需的计算资源也越多。
DFT计算的精度和计算时间取决于所使用的软件工具、参数和计算资源。在实践中,常常需要进行多次计算以确保得到准确的结果。为了提高计算效率,还可以使用一些加速技术,例如并行计算、GPU计算、预处理和近似方法等。
总结:第一性原理差分电荷密度计算是一种非常重要的计算化学方法,它可以提供关于分子和材料结构的详细信息,这些信息可以用于研究化学反应、物质性质和许多其他方面的研究。通过计算电荷密度函数,DFT可以提供一系列有用的信息,如能带结构、电子密度分布、光谱性质、化学反应和分子或材料性质等。在进行DFT计算时,需要选择适当的软件工具和参数,并使用加速技术来提高计算效率。
测试狗模拟计算