Single Gene Analysis Tool
简介:SGAT是一个免费开源的单基因分析工具,基于Linux系统实现自动化批量处理,能够快速准确的完成单基因和表型的关联分析,只需要输入基因型和表型原始数据,即可计算出显著关联的SNP位点,并自动生成结果报告。
安装与部署运行环境
-
官网渠道(推荐)
curl https://www.jewin.love/install.sh |sh
-
Github仓库
git clone https://github.com/JewinZao/SGAT.git
-
本地安装
wget https://www.jewin.love/SGAT-V1.1.0.zip
unzip SGAT-V1.1.0.zip
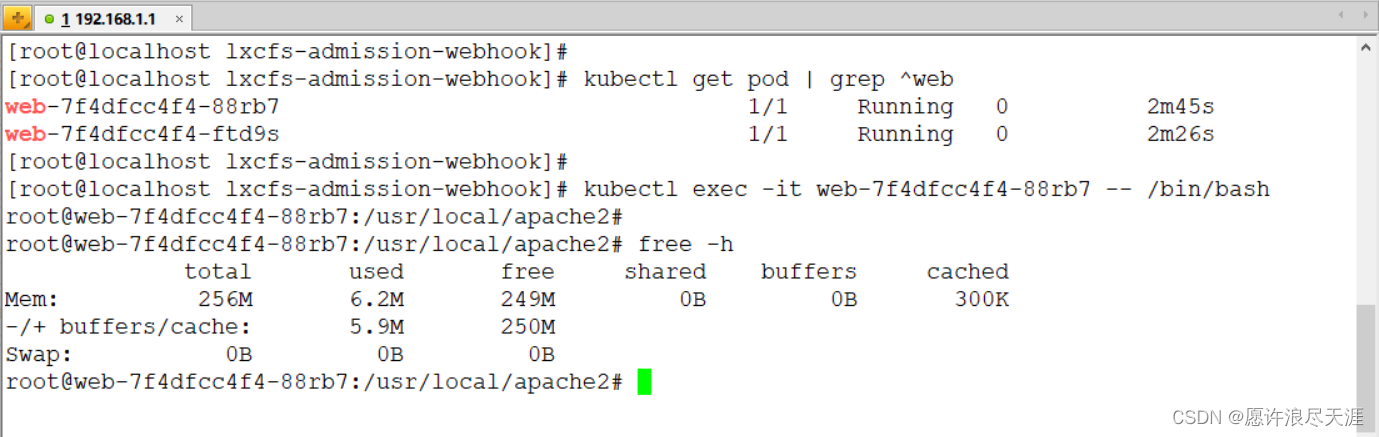
通过上述方式安装SGAT工具,安装完成后可以在当前目录下看到脚本文件即成功!
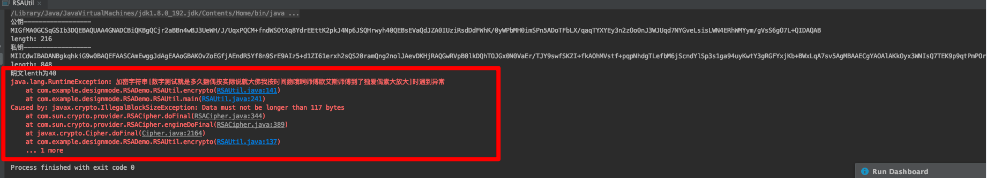
$ curl https://www.jewin.love/install.sh |sh
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 115 100 115 0 0 353 0 --:--:-- --:--:-- --:--:-- 353
--2023-04-15 16:25:20-- https://www.jewin.love/SGAT-V1.1.0.zip
Resolving www.jewin.love (www.jewin.love)... 101.34.79.245
Connecting to www.jewin.love (www.jewin.love)|101.34.79.245|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 371664 (363K) [application/zip]
Saving to: ‘SGAT-V1.1.0.zip’
100%[=================================================================>] 371,664 615KB/s in 0.6s
2023-04-15 16:25:21 (615 KB/s) - ‘SGAT-V1.1.0.zip’ saved [371664/371664]
Archive: SGAT-V1.1.0.zip
1090a66274055c0b2cc578a43f0a4bce083ede4b
Good finished!
依赖软件检查与安装
运行$ Rscript 1_check.R进行检查,根据提示安装相应软件和R包,直到所有依赖软件安装完成后提示finished,该过程也会自动检查基因型文件和表型文件,并对其进行提取,输出为列表,用于后续迭代计算。
###################### 单基因关联分析 ###########################
Design by Jewel
使用方法:
1.将所有的基因型文件放在02文件夹中
例如"TraesCS5A03G0123456.filter.vcf.gz"
2.将表型文件放在05文件夹中,命名为trait.txt
第一列名称为ID,后面每一列代表一个表型,例如"HT32L"
3.软件自动识别基因与表型信息
4.在当前文件夹下执行". ./start.sh"
5.结果将在后续生成
6.初始化与清除工作空间请执行". ./clearn.sh"
【 版本:V1.3.0 】
#################################################################
背景信息
什么是单基因关联分析?
单基因关联分析是一种遗传学和生物统计学方法,用于研究基因与特定表型之间的关系。在单基因关联分析中,通常比较来自不同群体的不同等位基因频率。如果某个等位基因在处理组中出现的频率显著高于对照组,则可以认为该等位基因与特定表型相关联。
单基因关联分析具有广泛应用,在医学、农业、动植物遗传学等领域都得到了广泛的应用!
待解决的问题
传统方式人工进行单基因关联分析需要从VCF文件开始,修改基因型文件,经过plink和taseel等软件转换文件格式,并手动修改变异信息,整理表型和基因型并互相匹配,逐步进行GWAS分析并根据结果作图,整个过程费时费力,而且极易出错。
因此,基于以上问题,开发了SGAT自动化单基因关联分析工具,快速完成多个基因多个表型多个模型的关联分析。
核心功能
-
变异信息自动识别与替换 -
染色体编号转换 -
基因型文件转换 -
表型与基因型匹配筛选 -
批量进行多模型GWAS分析 -
连锁不平衡作图 -
GWAS结果汇总整理 -
自动筛选显著性位点并提取变异信息 -
基因变异注释转换
定制化开发
-
GWAS分析模型自由选择 -
区间长度自由选择 -
筛选阈值自由选择 -
结果图片类型自由选择
源码开放性
Mar 29 22:55 0_README.md
Mar 22 20:25 1_check.R
Mar 19 21:40 2_gene_vcf2txt.R
Mar 22 20:12 3_hmp_trait_formate.R
Mar 20 11:05 4_GWAS_gapit.R
Mar 23 20:29 5_GWAS_results_translate.R
Mar 29 22:43 6_GWAS_Ttest_Result.R
Mar 19 21:39 chr_num2str.txt
Mar 22 20:14 clearn.sh
Mar 19 21:39 GAPIT1.txt
Mar 19 21:39 GAPIT2.txt
Mar 19 21:39 GAPIT_install.R
Mar 19 21:39 GAPIT.R
Mar 31 11:53 start.sh
上述所有源码均在Github存放,其中bash脚本clearn.sh的功能是初始化工作目录并清除临时数据,start.sh的功能是启动自动化进程。之后将会在公众号“生信分析笔记”上进行原理与算法介绍,欢迎提交遇到的BUG到https://github.com/JewinZao/SGAT/issues
参考资料:
Plink、Tassel、LDBlockshow、GAPIT、Tidyverse、vcfR、ape、do、multtest、LDheatmap、genetics、scatterplot3d、EMMREML等
声明:
SGAT遵循国际GNU General Public License v3.0,核心算法和代码均开源公布,进行科学研究学习交流,不涉及商业使用,如果有任何问题欢迎联系。
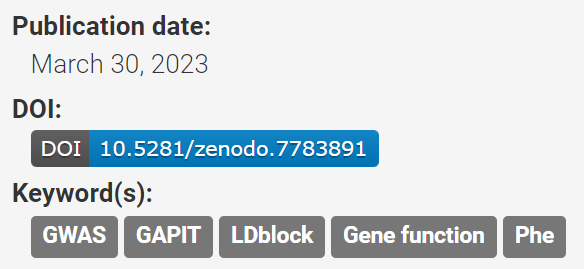
软件公开发布链接:
https://doi.org/10.5281/zenodo.7783891 
本文由 mdnice 多平台发布