MMPBSA计算–基于李继存老师gmx_mmpbsa脚本
前期准备
软件安装
安装gromacs, 可以查阅 我的blogGromacs-2022 GPU-CUDA加速版 unbantu
安装 apbs, sudo apt install apbs
安装 gawk, sudo apt install gawk
MD模拟好的文件
我们以研究蛋白小分子动态相互作用-III(蛋白配体复合物 GROMACS)模拟好的文件为例子
需要准备如下文件
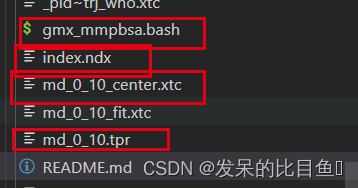
脚本执行
MD 做完后需要修复轨迹
gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_center.xtc -center -pbc mol -ur compact
采集最后5ns的模拟数据
gmx trjconv -f md_0_10_center.xtc -o trj_5-10ns.xtc -b 5000 -e 10000
检查轨迹路径
gmx check -f trj_5-10ns.xtc
检查index.ndx, 配体的索引只能是一个,多的必须删除

下载 gmx_mmpbsa.bash,网址:https://github.com/Jerkwin/gmxtools/blob/master/gmx_mmpbsa/gmx_mmpbsa.bsh
修改文件:

修改为:
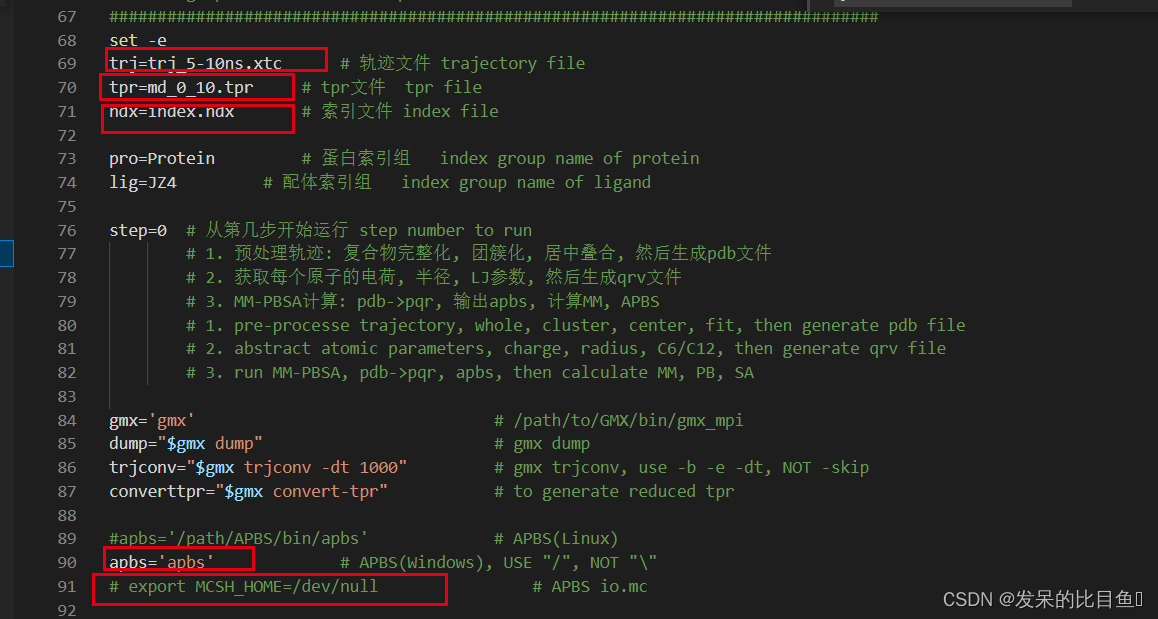
执行文件:
bash gmx_mmpbsa.bash
最终结果
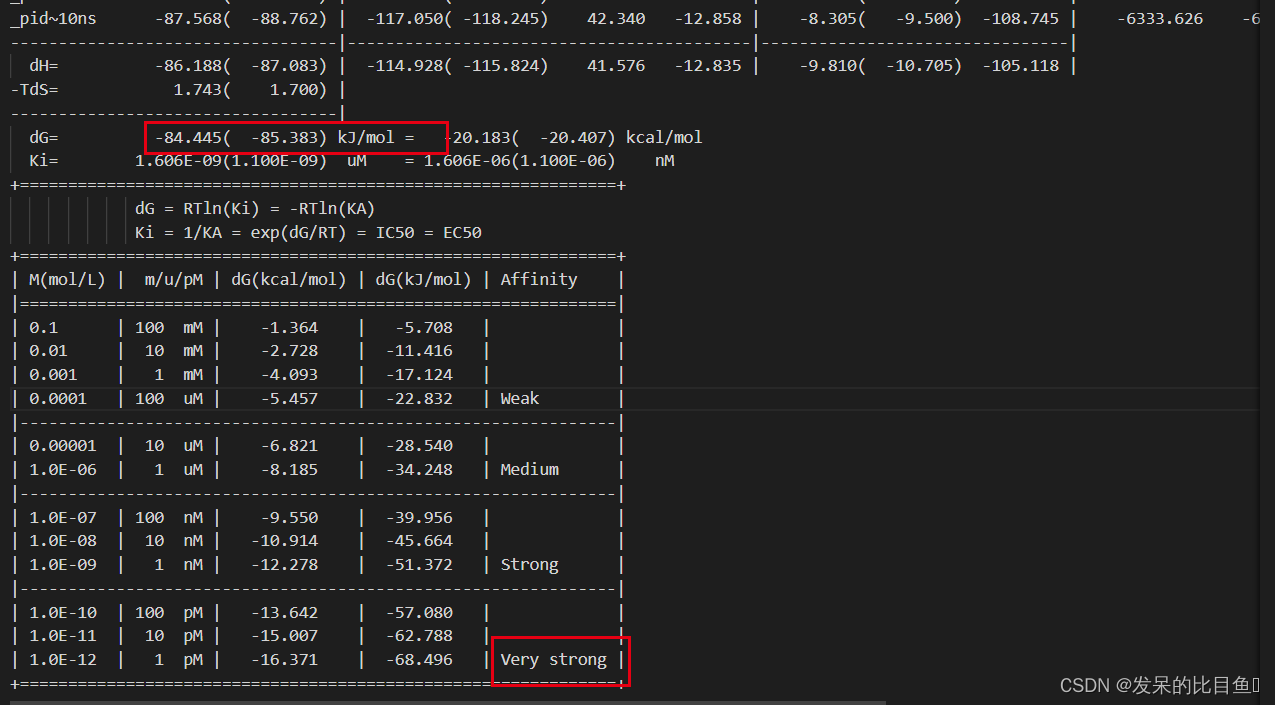
结果结合力非常强!!