The evolutionary tale of lilies: Giant genomes derived from transposon insertions and polyploidization
百合的进化故事:由转座子插入和多倍化导致的巨型基因组

百合(Lilium spp.),被誉为“球根花卉之王”,因其美丽芳香的花朵和富含营养的鳞茎而具有极高的观赏和药用价值。百合属包含约 115 种多年生球根草本植物,广泛分布于北半球,其中中国已发现 55 种和 18 个变种。中国丰富的百合种质资源不仅促进了其广泛应用,也为科学研究提供了宝贵的材料。然而,百合的巨型基因组对高质量基因组组装提出了挑战,目前尚无该类群的参考基因组。迄今为止,由于技术限制和高昂成本,仅有少数巨型基因组被成功解析。
生物体基因组大小的巨大差异在生物学上具有重要意义,但生物复杂性与基因组大小之间的关系仍不明确。大规模和复杂基因组的测序与组装面临诸多挑战,如多倍化、高杂合度和高重复序列比例。由于百合属植物的基因组比其他真核生物大得多,几乎所有百合种都可作为研究生物复杂性与基因组大小关系的理想模型。兰州百合(L. davidii var. unicolor)是唯一可食用的甜百合品种,在中国兰州已被种植约 150 年,为当地农民提供了重要的经济来源。我们选择该品种进行基因组测序,以获取有价值的数据来研究百合的巨型基因组,从而推动百合的遗传改良和育种工作。
百合巨型基因组的染色体级组装
我们通过流式细胞术分析估算 L. davidii var. unicolor 的基因组大小约为 38.01 Gb,而 K-mer 分析结果显示其基因组大小约为 37.62 Gb,杂合度为 2.18%。核型分析表明该品种为二倍体,具有 12 对巨型染色体。为了组装这一超大基因组,我们生成了 3.32 Tb 的 Illumina 短读长数据和 2.25 Tb 的 Nanopore 长读长数据,并采用适用于巨型基因组的优化 NextDenovo 流程,构建了初步的非冗余 contig 级组装,得到 36.68 Gb 的基因组序列(13,068 个 contigs,N50 = 7.72 Mb)。随后,我们生成了 4.45 Tb 的高通量染色体构象捕获(Hi-C)数据用于物理图谱构建,并经过四轮手动调整,将 96.99% 的 contigs 锚定到 12 条拟染色体上,对应 12 对染色体(图 1A)。

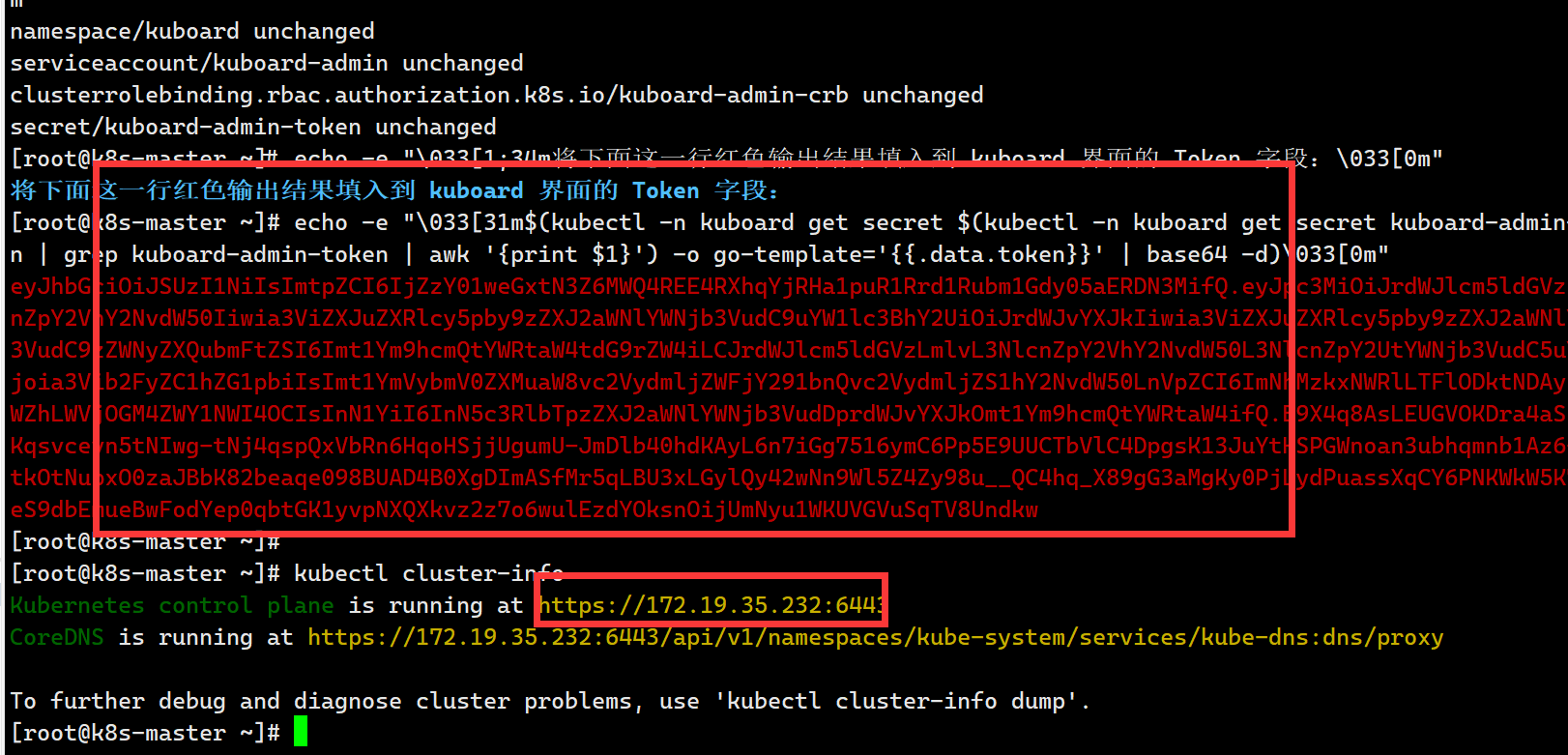
图 1. 百合基因组及多组学分析
(A) L. davidii var. unicolor 的基因组特征。 (B) Gypsy 和 Copia 类逆转录转座子 (retrotransposons) 的插入时间估算。 (C) Gypsy 和 Copia 逆转录转座子亚类的系统发生关系及分类。 (D) 全基因组复制 (WGD) 事件的鉴定。 (E) 百合的系统发生位置及分化时间。“τ” 代表所有单子叶植物(除水龙骨目和泽泻目外)共享的 WGD 事件。 (F) 百合基因组中不同基因长度和内含子数量的表达模式。 (G) 百合小鳞茎发育过程中鲜重及淀粉和蔗糖含量的变化。 (H) 糖酵解代谢物与共表达模块的相关性分析。
我们采用多种方法评估了基因组组装的完整性和准确性。首先,我们测量了三个体细胞中 12 对染色体的相对物理长度,结果显示组装的染色体长度与实际观测的物理长度成比例。随后,我们将 Illumina 和 Nanopore 读段重新比对至基因组,映射率分别高达 97.80% 和 99.10%。长末端重复 (LTR) 组装指数 (LAI) 评估值超过 10,而碱基水平的准确性分析得出的质量值 (QV) 为 30.18,远超参考级基因组的标准。此外,我们在两个拟染色体的两端以及五个拟染色体的一端鉴定到了端粒序列,进一步验证了基因组组装的高完整性。基因组注释共鉴定到 87,501 个蛋白编码基因,其中 78,348 个基因(89.54%)可进行功能注释。此外,通用单拷贝直系同源基因 (BUSCO) 评估结果显示基因组完整性达 94.90%。这些结果表明百合基因组的完整性、准确性和连贯性均处于较高水平。
近期转座子扩增及多倍化导致兰州百合的超大基因组
基因组大小的主要影响因素包括重复 DNA 序列的积累和全基因组复制 (WGD)。注释分析表明,百合基因组中重复序列比例极高 (88.31%),其中转座元件 (TEs) 占比 84.19%。在这些 TE 中,LTR 逆转录转座子 (LTR-RTs) 是主要组成部分 (64.40%),其中 Copia 和 Gypsy 元素分别占 16.62% 和 31.53%。LTR-RTs 插入时间估算结果显示,在过去 500 万年内,其积累速率显著加快,其中 Copia 在约 165 万年前达到峰值,出现爆发式插入,而 Gypsy 在约 89 万年前出现插入高峰 (图 1B)。在这些高峰时期,Copia 和 Gypsy 元素的插入分别占 TE 总插入量的 29.6% 和 22.1%。因此,近期的大规模 TE 插入事件是百合基因组膨胀的主要原因。
系统发生分析表明,百合中的 Gypsy 和 Copia LTR-RTs 均包含多个亚类 (图 1C)。其中,两个 Gypsy 亚类 (Athila 和 Tekay) 以及两个 Copia 亚类 (Tork 和 Ikeros) 的丰度远高于其他亚类,表明它们对基因组扩张贡献显著。在植物进化过程中,不同 TE 亚类扩增程度各异,而在百合中,Athila、Retand、Tekay 和 Tork 发生了爆发式扩增 (图 1C)。部分亚类(如 Athila)具有着丝粒周围异染色质区域的靶向偏好,可抑制重组,从而降低 LTR-RT 的移除率,进一步促进基因组扩张。
除了 TE 插入,全基因组复制 (WGD) 也可能直接导致基因组大小的扩增。因此,我们分析了百合基因组内的 WGD 事件。对保留在共线性区域内的直系同源基因进行同义替换位点 (Ks) 分布分析,结果显示两个显著峰值 (Ks = 1.35 和 0.97),表明百合经历了两轮 WGD 事件 (图 1D)。跨基因组共线性分析显示,百合与 Acorus gramineus 之间的共线性比例为 4:2,与芦笋 (Asparagus officinalis) 和山药 (Dioscorea alata) 之间的比例均为 4:4。鉴于 A. gramineus 仅经历了一次 WGD,而芦笋和山药各经历了两次 WGD,这些共线性关系支持百合基因组中发生了两轮 WGD 的假设。
使用 563 个低拷贝直系同源基因构建的单子叶植物系统发生树显示,百合是天门冬目 (Asparagales) 的姐妹类群,二者在约 72.7 百万年前 (mya) 分化 (图 1E),这一结果与大多数基于核基因的研究一致。然而,一些基于叶绿体基因组的研究提出天门冬目和百合目 (Liliales) 依次与鸭跖草类群 (Commelinids) 形成姐妹关系,可能反映了不同遗传方式(双亲遗传 vs. 单亲遗传)导致的核质冲突。此外,尽管洋葱和大蒜等天门冬目植物经历了两次额外的 WGD,但其基因组大小仍不足百合基因组的一半,说明百合在 TE 插入和 WGD 事件后,基因组得到了更好的保留。LTR-RTs 的不等重组是植物中主要的 TE 移除机制,而百合可能比其他植物拥有更低的不等重组率,从而导致其 LTR 积累速度更快,形成超大基因组。
百合的超长基因源自超长内含子,并表现出基因长度与表达水平的负相关性
百合基因组中包含大量长基因,平均长度为 57.61 kb,长度超过 50 kb 的超长基因占所有基因的 33.88%。然而,百合的编码序列 (CDS) 平均长度仅 847.17 bp,包含平均 3.97 个外显子,每个外显子的长度为 213.72 bp,表明超长内含子是百合超长基因的主要原因。百合基因的平均内含子长度达 ∼19.13 kb,在已发表的植物基因组中仅次于攀枝花苏铁 (Cycas panzhihuaensis)。
在裸子植物华山松 (Pinus tabuliformis) 中,长内含子基因的表达水平较高,被认为与染色质可及性有关。然而,百合基因组的表达模式显示出不同的趋势 (图 1F):在基因长度小于 50 kb 时,基因长度与表达水平呈正相关,而当基因长度超过 50 kb 后,这种相关性变为负相关。此外,表达水平最高的百合基因通常包含 3 或 4 个内含子 (图 1F)。这些发现表明 50 kb 可能是影响基因转录或内含子剪切效率的关键阈值。
百合小鳞茎发育与碳水化合物代谢
百合鳞茎是重要的营养储存器官,在东亚的医药和食品产业中具有重要价值。为探究小鳞茎发育过程中营养物质的积累及其分子机制,我们对五个发育阶段的小鳞茎进行了细胞学、转录组学和代谢组学分析。整个发育过程中,小鳞茎中的淀粉和蔗糖含量逐步积累 (图 1G)。代谢-转录组相关性分析显示,碳水化合物代谢物与一个基因表达模块(turquoise)存在显著相关性 (图 1H),该模块可能包含碳水化合物和淀粉代谢途径中的关键基因。
总之,本研究成功组装了染色体级百合基因组,并深入解析了其巨型基因组特征,为百合遗传改良和分子育种提供了重要资源。








![[实现Rpc] 服务端 | RpcRouter实现 | Builder模式](https://i-blog.csdnimg.cn/img_convert/b310cdcebdee1cee2ff29c6a79b50de0.png)