1
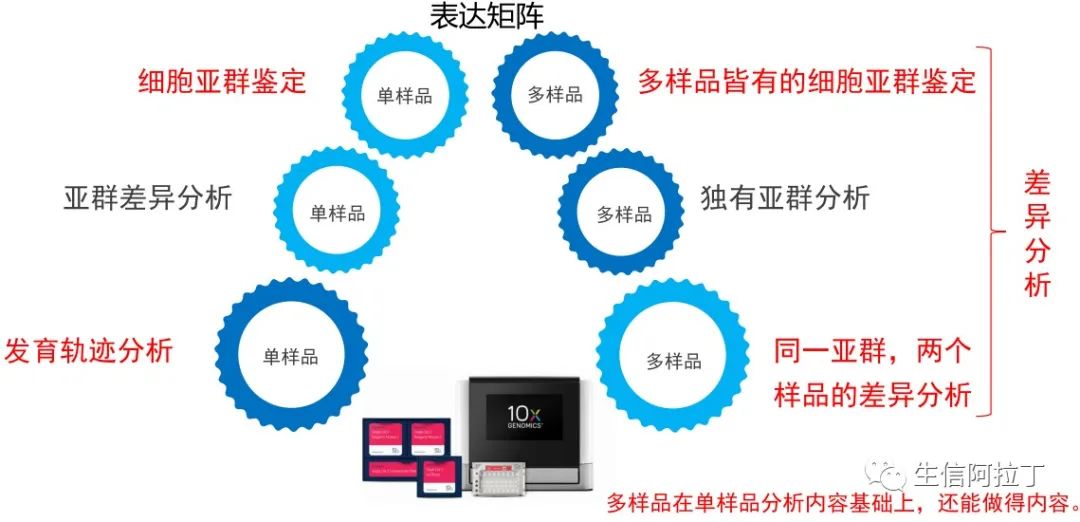
单细胞转录组亚群常见分析内容
· 重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)
· 如何使用Bioconductor进行单细胞分析?
单细胞转录组亚群分析的内容根据样品数目多少,可以分为单个样品或者多个样品。
单个样品主要可以进行的分析内容有:细胞亚群的鉴定(Celaref | 单细胞测序细胞类型注释工具)、亚群之间的差异以及发育轨迹分析(NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集)。多个样品分析内容包括所有单个的分析内容,并且在此基础上还能进行样品的差异分析。
这里样品差异分析主要分两个方面:
1. 从宏观上来说,不同亚群中不同样品的细胞数目的差异,不同亚群细胞具有不同的功能,因此亚群的差异对于研究异质性具有十分重要的作用。
2. 从单个亚群来说,可以研究不同样品之间的差异,比如同样是上皮细胞,我们可以研究上皮细胞中不同样品之间的差异,基因表达或者代谢通路的差异,这是从机理上来解释生物学问题。
可以使用Seurat工具进行细胞亚群分析(单细胞分析Seurat使用相关的10个问题答疑精选!)。
链接:
https://github.com/satijalab/seurat
2
数 据 质 控
1
空载细胞、双细胞、多细胞数据质控
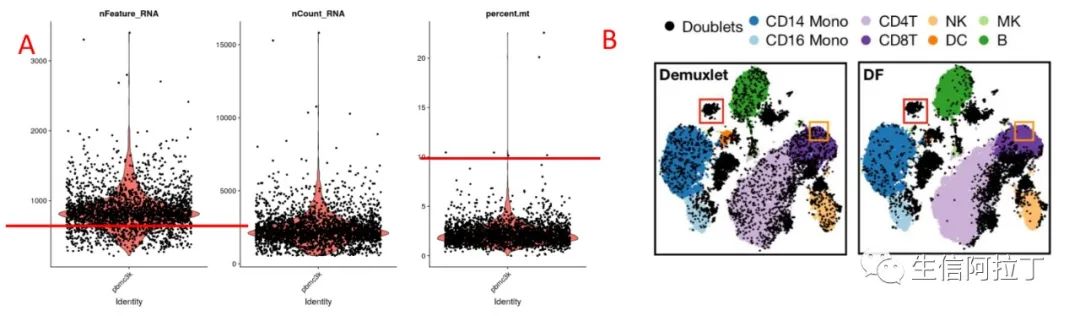
如上图A左图所示,为每个细胞的基因表达数目小提琴图(可视化之为什么要使用箱线图?)。一般对于单细胞转录组来说,如果细胞表达的基因数目过少,可能是空载细胞(在细胞分筛的时候,未捕获到细胞,只是捕获了溶液中的游离mRNA);如果细胞基因数目表达过高,可能是同时捕获2个或多个细胞,统一命名为双细胞(2个以上的细胞的基因表达数目一般就会较高)(单细胞预测Doublets软件包汇总-过渡态细胞是真的吗?)。
图A右图为每个细胞中线粒体基因(线粒体基因的名称一般为mt开头,不同物种大小写可能不一样)的表达UMI占比,除非特别的样品或者组织(比如卵组织、心脏、癌细胞等),一般细胞的线粒体基因表达占比较低。不同文献会有不同的阈值,5-40%都有,因此在做此数据质控的时候需要根据自己的研究样品设定一个合适的阈值,通常可以设为20-25% (对一篇单细胞RNA综述的评述:细胞和基因质控参数的选择)。
图B为双细胞检测方法介绍,如果细胞基因的表达数目过高,可能对结果具有较大的影响(比如有时过渡态细胞可能不是过渡态细胞,而是双细胞),因此一般需要注意双细胞数目。一般来说,10xGenomics单细胞转录组平台对双细胞有一定的控制,1000个细胞双细胞率不超过0.9%,10000个细胞不超过7%。但是有时候由于实验因素,可能会偏高,需要在分析的时候去掉双细胞。
过滤双细胞的方法有很多种,一种比较直接粗暴的方法就是把细胞基因表达数目超过一定阈值的细胞去掉(比如PBMC细胞,阈值为2500),不过不同的样品阈值不同;另外一种方式就是通过算法来去掉双细胞,现在去掉双细胞的工具有很多种,比如:
DoubletFinder :https://www.cell.com/cell-systems/fulltext/S2405-4712(19)30073-0、
scrublet :https://github.com/AllonKleinLab/scrublet
DoubletDecon:https://github.com/EDePasquale/DoubletDecon
DoubletDetection:https://github.com/JonathanShor/DoubletDetection.git
等等,python和R语言的工具都有,可以根据自己需要进行工具选择。
对于空载细胞分析,一般cellranger流程已经进行处理过。cellranger(10X单细胞测序分析软件:Cell ranger,从拆库到定量)在进行call cells的时候,会通过EmptyDrops工具根据其表达量与背景表达量的相似性进行空载细胞的判断。链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1662-y
当然也会对基因进行质控,一般会过滤掉基因表达细胞数目过低的基因,比如一般要求至少在3多个或者5个细胞表达的基因(对一篇单细胞RNA综述的评述:细胞和基因质控参数的选择)。
2
特定细胞质控
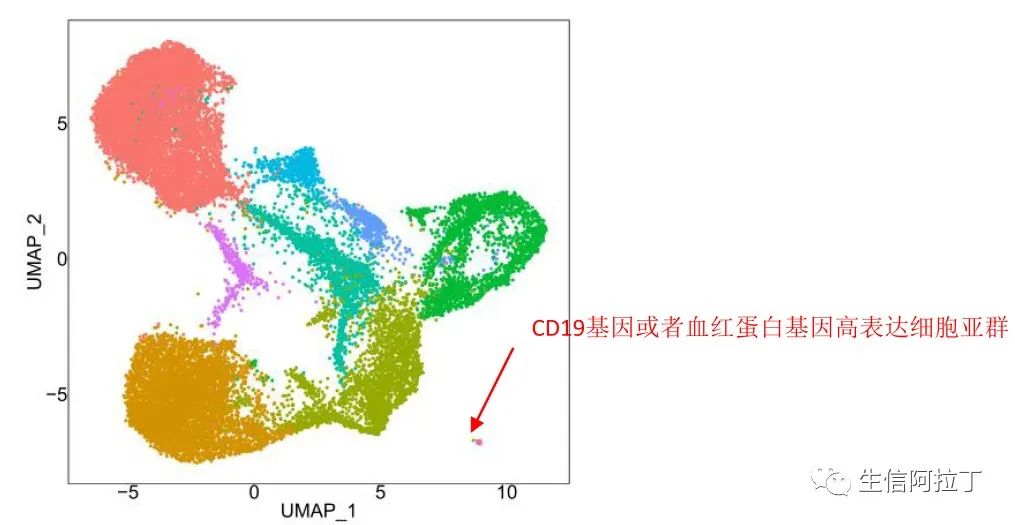
特定细胞的质控,这个一般需要通过特定的样品进行分析,比如PBMC细胞中不能含有血红蛋白基因高表达的细胞,血红蛋白基因高表达细胞一般是红细胞,但是红细胞对于我们的研究一般来说没有什么意义,因此一般需要过滤掉此类细胞,这个阈值在不同的样品设定的不一样,比如可以设定阈值为5%。
其他特定细胞的质控,比如我们经过流式筛选的是T细胞,结果出现高表达B细胞、巨噬细胞等其他细胞类型的marker基因,因此需要去掉此类细胞,一般此类细胞聚类都分离得比较开,与其他细胞不一样,比如下图所示。
3
降维可变基因和PCA数目选择

如上图所示,一般会根据Mean-variance选择变化度较大的基因进行后续降维聚类分析。一般选择top1000或者2000基因进行PCA降维。然后聚类时,一般是挑选特定的PCA数目进行聚类,比如图B所示的碎石图,一般会选择在拐点位置的PCA数目进行后续分析。
4
细胞周期质控
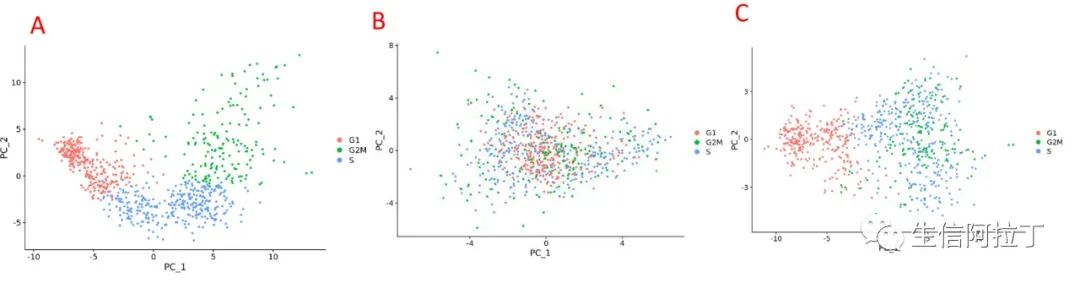
上述图A为没有进行细胞周期(Seurat亮点之细胞周期评分和回归)变异的排除,图B为进行细胞周期变异的排除,图C为S.Score-G2M.Score进行变异的排除的结果。目前主要是人、鼠有细胞周期基因,如果想对其他物种进行细胞周期分析,可以根据与人的同源基因比对后进行(OrthoMCL鉴定物种同源基因 (安装+使用))。
另外需要注意的是,不是所有的样品都适合进行细胞周期分析,如果是干细胞分化研究,这样的话就可能有点不合适,使用两者之间的差异进行分析会更合适一些。具体情况需要根据样品进行分析(这样会将细胞周期细胞与非周期细胞分隔开)。
3
细 胞 亚 群 差 异
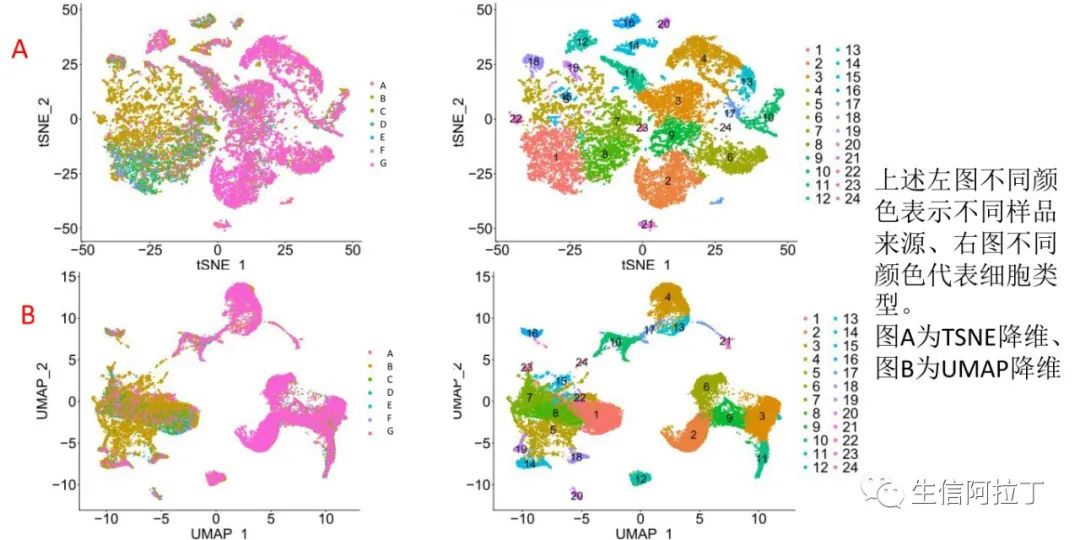
在进行所有质控后,进行降维聚类,得到如上的聚类结果图。现在降维可视化的方法主要有TSNE和UMAP,效果都比较好,可以都绘制出来看下哪个更适合自己的数据(Hemberg-lab单细胞转录组数据分析(十二)- Scater单细胞表达谱tSNE可视化) (还在用PCA降维?快学学大牛最爱的t-SNE算法吧, 附Python/R代码)。
但是聚类的结果与可视化的方法没有任何关系,比如上图,虽然图A和图B图形不一样,但是其聚类结果完全一致。 这是个很有意思的问题,有意愿的我们留言讨论,当然生信宝典往期文章也有解答。
4
亚 群 细 分 策 略
一般单细胞转录组很少能够一次性得到符合预期的结果,需要对结果需要调整,比如需要对亚群数目进行调整。
如果使用Seurat (https://satijalab.org/seurat/)工具的话,可以通过调整FindClusters函数的resolution参数进行调整,一般可以设0.1-1之间,这个值越高,得到亚群数目越多,但是细胞亚群数目不能太多,不然后续分析比较耗费精力。
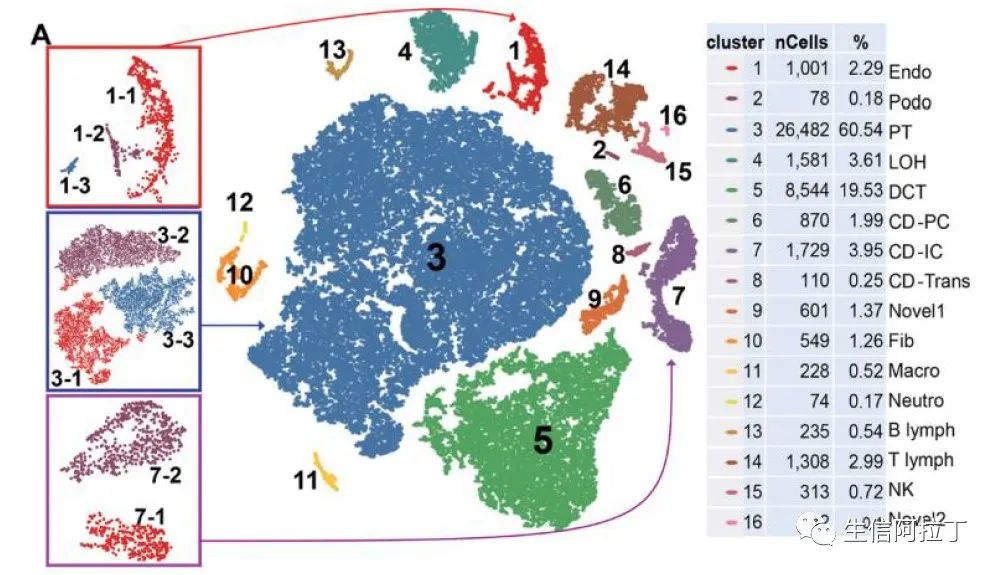
另外一种需要进一步或者调整结果的是,对感兴趣的细胞亚群进行亚群细分,可以把感兴趣的一个亚群或者多个亚群提取出来,然后再进行亚群细分。
亚群细分一般有两种方式:第一种,通过分辨率,可以使亚群数目增多。比如下图的1亚群,可以看到1亚群和1亚群的细分的图形一致;第二种,将此亚群提取出来,然后再整体的按照之前分析的pipeline进行重新分析。因此其亚群细分的结果图形会发生变化,比如下图的7亚群细分。
5
亚 群 功 能 分 析
一般进行简单细胞亚群分析后,会再对亚群进行差异分析。进行差异分析时,一般是选择该亚群与除了该亚群以外所有的亚群进行分析,一般阈值有pct(亚群中某个基因表达的占比)和差异倍数(平均表达量),对于Seurat 工具来说,差异倍数一般设为0.25,pct阈值一般是该亚群和该亚群以外所有亚群至少一个的pct值大于0.1。通过此方法得到的差异基因,也认为是marker基因,即每个亚群特异的基因。
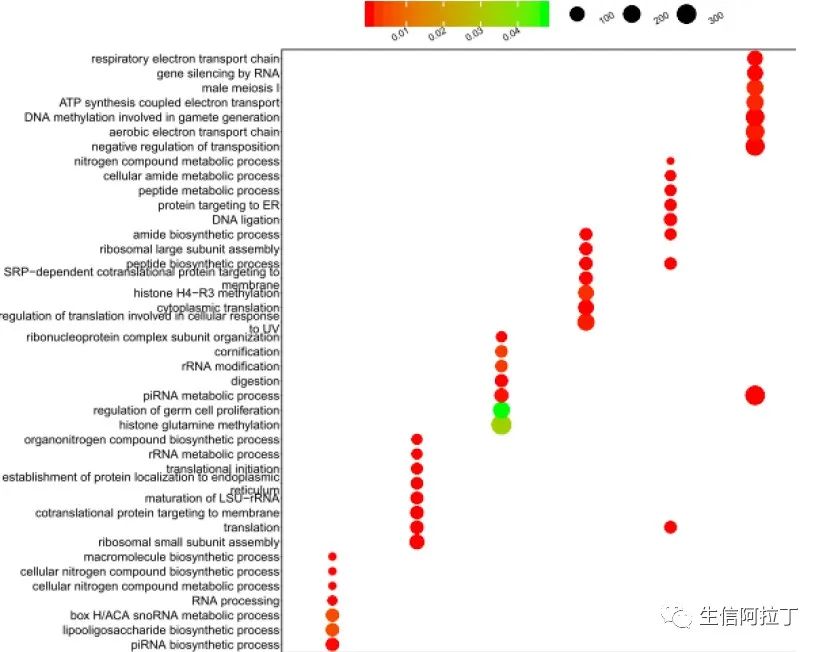
根据上述方法得到的差异基因,进行功能分析,了解每个亚群特异的功能,一般会进行GO(GO、GSEA富集分析一网打进)和KEGG分析(Pathview包:整合表达谱数据可视化KEGG通路),然后通过气泡图展示差异,如下图所示:
6
细 胞 亚 群 鉴 定
根据上述得到的marker基因,对细胞亚群进行鉴定,这也是单细胞转录组分析最重要的一步,也是最关键的一步,通常需要花费大量的精力进行细胞亚群鉴定。
通常细胞亚群鉴定的方式有如下四种:传统经典marker基因、自动化鉴定工具、其他单细胞转录组数据映射、与bulk RNA相关性分析。
01
传统经典marker基因
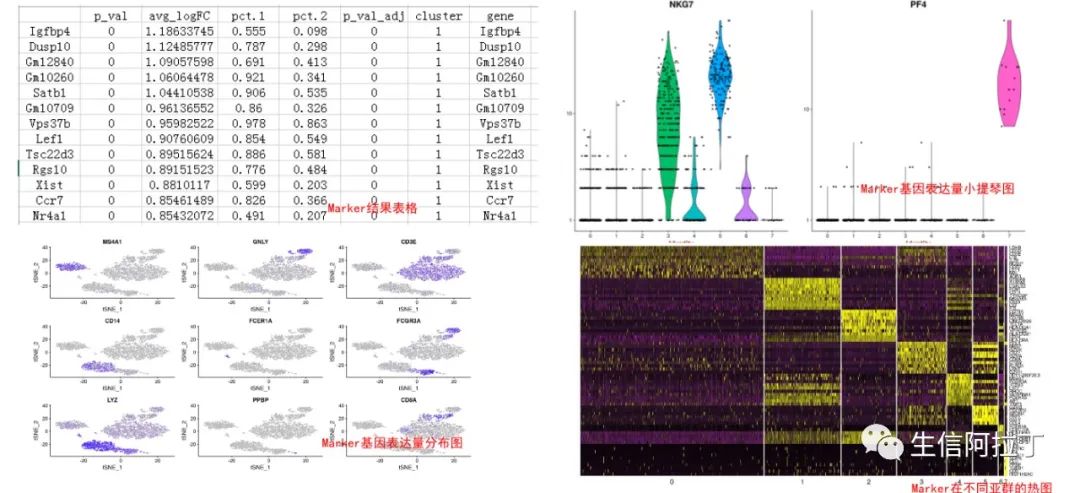
根据已知的细胞类型的marker基因进行细胞亚群鉴定(单细胞分群后,怎么找到Marker基因定义每一类群?)。如上图右上角小提琴图,可以明显看出PF4基因在亚群7特异性表达,因此可以根据此基因为某些细胞类型marker基因进行细胞亚群鉴定。
一般亚群鉴定不是单独一个基因,可能需要多个基因。说到这里,我们需要知道传统经典的marker基因,这个表格从哪儿来呢?
一般有如下两个常用数据库:
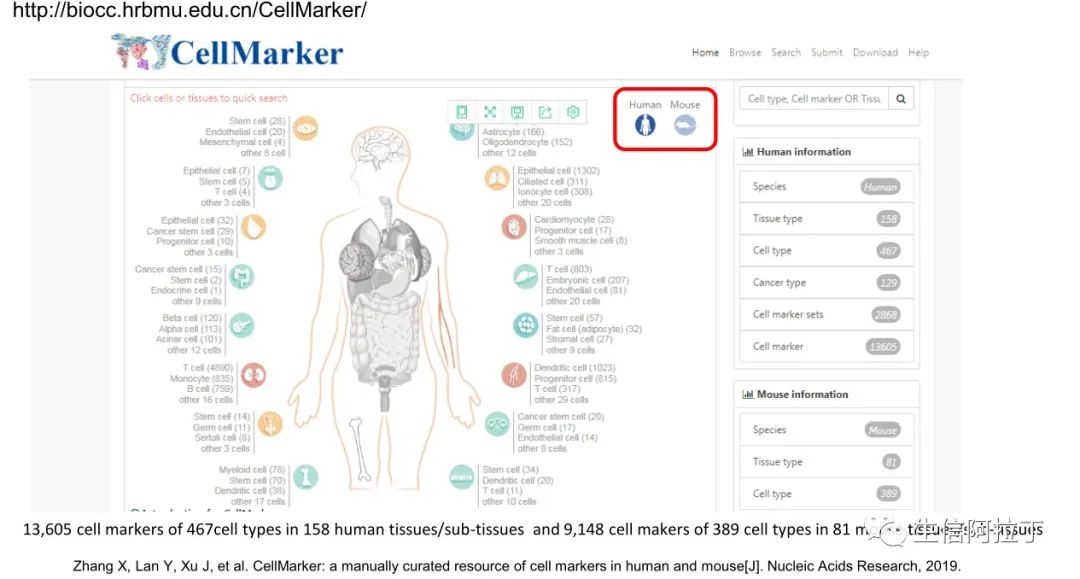
CellMarker:(单细胞分群后,怎么找到Marker基因定义每一类群?)
http://biocc.hrbmu.edu.cn/CellMarker/
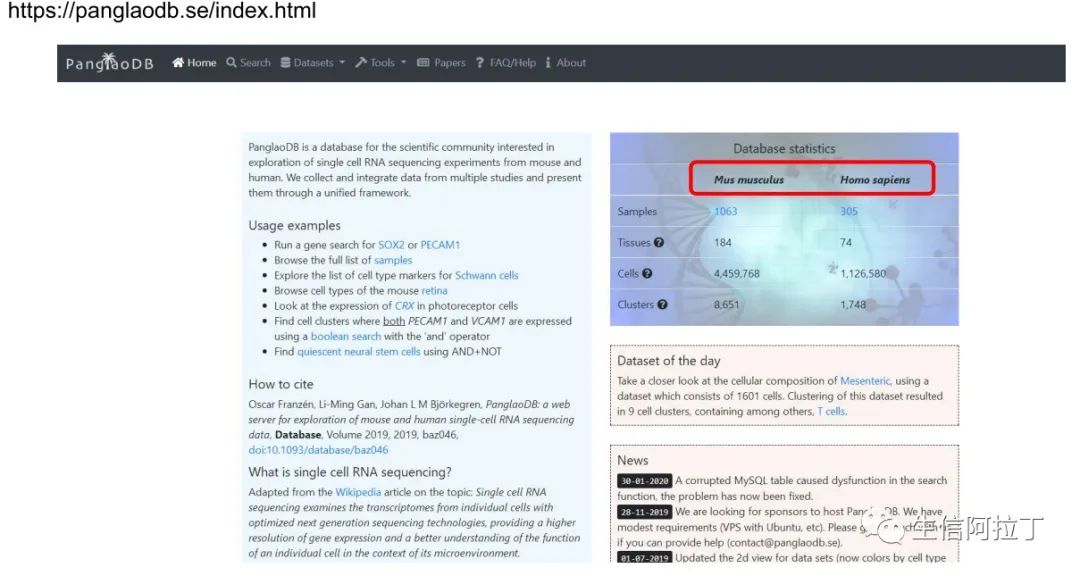
panglaodb:
https://panglaodb.se/index.html
不过这两个数据库都只是提供了人和小鼠相关的数据,没有其他物种的,因此其他物种最好通过查询相应的文献来确定。
02
自动化鉴定工具
目前单细胞亚群鉴定的自动化工具有很多种,至少有20-30种,这些工具主要有两种,一种是自动化鉴定,另外一种是半监督的方式。
自动化鉴定比较常见的singleR,内置了人和小鼠的数据,其基本原理是通过计算单细胞与内置数据库的相关性来判断细胞类型,也可以自己建数据库(七龙珠|召唤一份单细胞数据库汇总)。地址为:
https://github.com/dviraran/SingleR
优点是不用自己提供细胞类型以及相应的marker基因,但是其缺点是只能鉴定出数据库已有的细胞类型以及不能鉴定特别细的细胞亚群,特别细的细胞亚群比如CD4+T 细胞亚群再细分,就没法完成了。

另外一种是半监督的方式,需要自己提供细胞类型的marker基因,也就是只能鉴定自己提供的细胞类型,一是限制了细胞类型,另一方面则是可以鉴定任意感兴趣的细胞类型,不过这种方式需要老师具有较深厚的生物学背景。
比较常用的软件有cellassign 和Garnett,其中cellassign(cellassign:用于肿瘤微环境分析的单细胞注释工具(9月Nature))只要提供细胞类型以及对应的基因,软件根据TensorFlow机器学习的方法,对每个细胞进行打分。Garnett是拟时间分析工具-monocle工具编写的团队开发的一种细胞简单快速注释细胞类型的工具。它不仅仅提供了根据基因鉴定细胞亚群,还可以通过设定基因表达量阈值、或者不含某个基因来筛选,而且官方提供了一定数目的细胞类型的marker基因list。
cellassign地址:
https://irrationone.github.io/cellassign/index.html)
Garnett地址:
https://cole-trapnell-lab.github.io/garnett/)
03
不同单细胞数据映射
其实现在很多人都会问,现在已经有那么多单细胞转录组数据,为什么不可以利用已知的单细胞数据来鉴定未知单细胞数据?
其实是可以的,而且这种操作的方法和工具还挺多的,比较常用的是Seurat工具中有个TransferData 函数,可以将别的数据标签映射到未知的数据,从而鉴定细胞类型,测试数据准确性可达90%以上。地址:
https://satijalab.org/seurat/v3.1/integration.html

04
与Bulk RNA数据进行相似性分析
另外一种比较常见细胞亚群鉴定方式就是,用单细胞属于相应细胞类型的细胞系测序获得的Bulk RNA数据进行相关性分析,得到相似性热图(这也太简单了吧!一个函数完成数据相关性热图计算和展示),判断细胞类型,结果如下图。
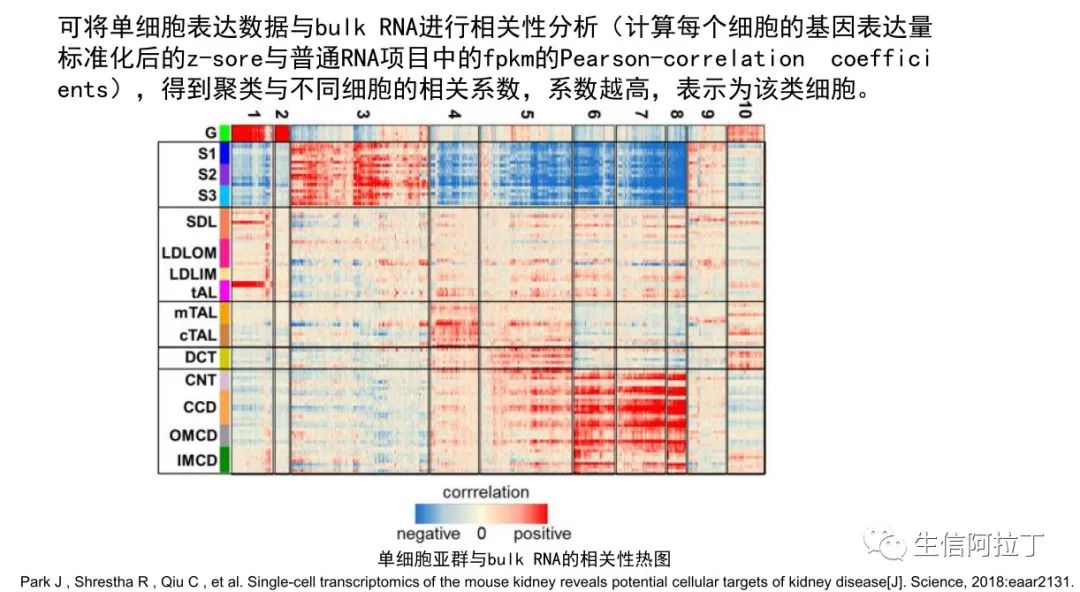
此方法是比较耗费精力,需要收集相应细胞类型的Bulk RNA数据,另外单细胞数据表达的模式可能与Bulk RNA数据不太一致,因此此方法一般用于数据验证,不作为鉴定结果。
最后说一句,其实也有一些常用的富集分析工具,通过对输入的marker基因进行富集(拿到基因两眼一抹黑?没关系,先做个基因富集分析吧!),得到可能细胞亚群,不过有个缺点就是,对于一些通过流式得到的单细胞转录组数据,比如某个细胞只有CD4+和CD8+细胞,可能CD4+和CD8+基因不是marker基因,不在marker基因list中,因此鉴定会有些问题。
7
单细胞转录组样品差异分析
10xGenomics单细胞转录组一次可获取10000个细胞。在研究的时候,不能一个细胞一个细胞研究,一般通过降维聚类,将表达模式相同的细胞聚类在一起,即得到细胞亚群,随后的研究是基于细胞亚群进行。
研究不同样品、不同处理条件、不同组织样品的时候,一般是在同一个亚群之下进行的研究,毕竟不同类型的细胞其表达模式肯定不一样,这个可比性不太强。比如在使用某种药物处理后,想看看CD4+ naive T细胞有什么变化,然后挑取CD4+ naive T细胞亚群,直接比较两个样品的细胞基因表达差异,然后对此基因进行功能注释,了解哪些关键基因发生变化,代谢通路发生了变化。
由于10xGenomics单细胞转录组的基因表达较smart-seq2的数据较低,因此一般进行差异分析的时候,其阈值不能直接按照smart-seq2的阈值设置,Seurat一般设为差异倍数的log值为0.25。(什么?你做的差异基因方法不合适?)
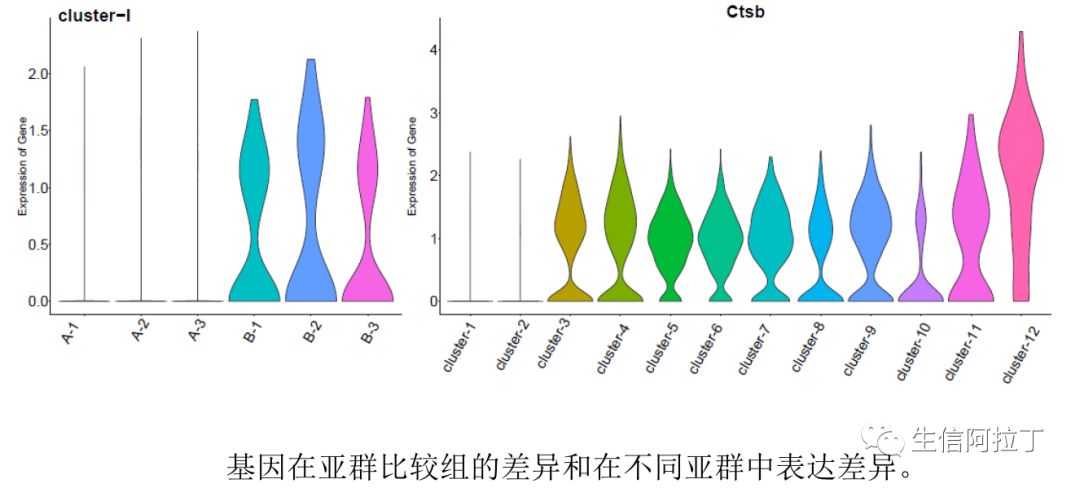
我们也可以根据每个样品在某个亚群中所有细胞基因的平均表达量作散点图,这样能更直观地展示差异基因,比如下图展示了top10基因,一般越靠近y轴的基因,就是STIM高表达,越靠近x轴,就是CTRL高表达。下图使用了不同的差异基因展示方式。
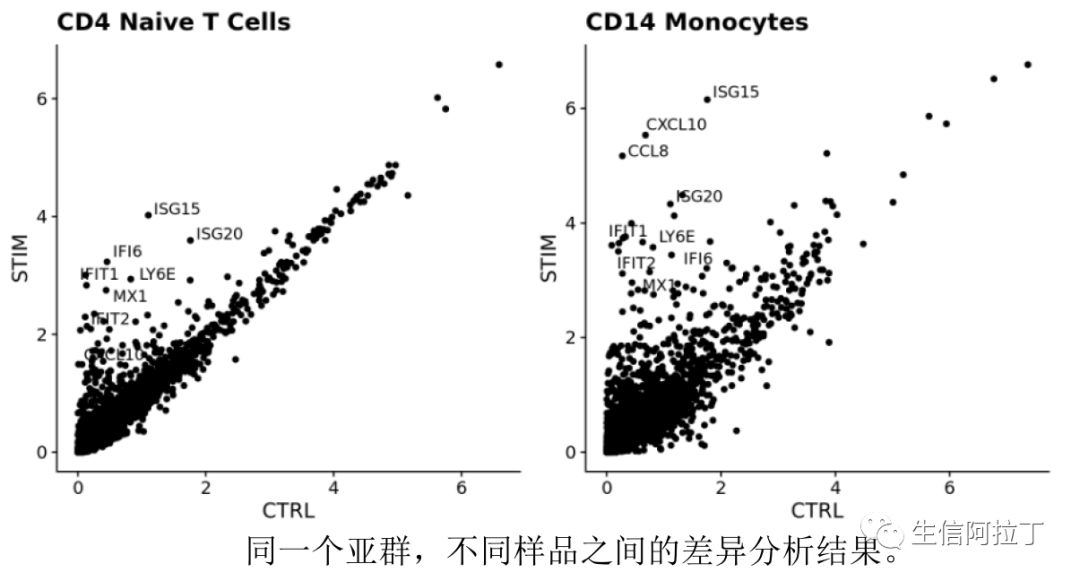
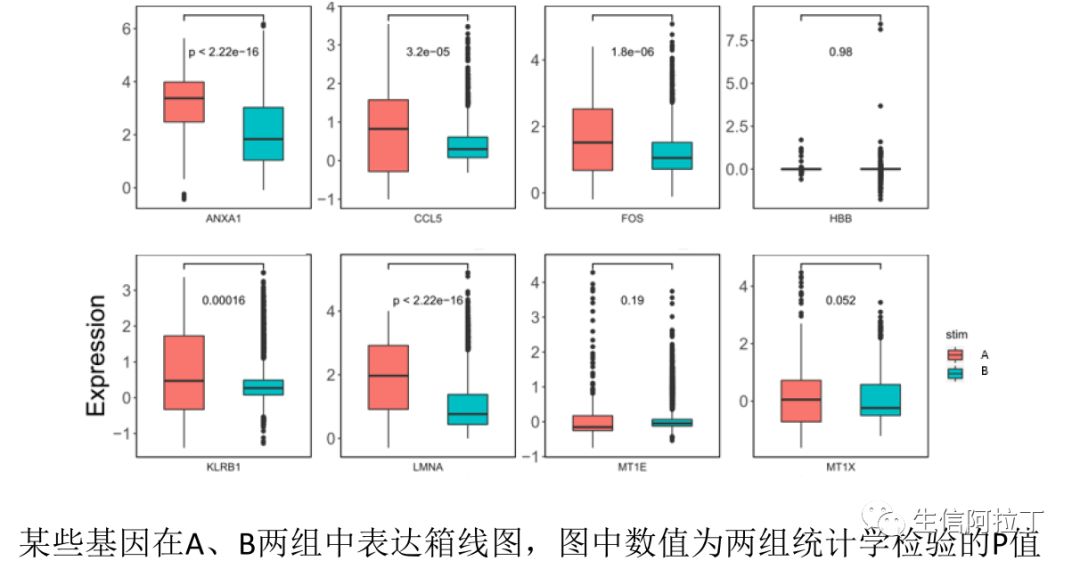
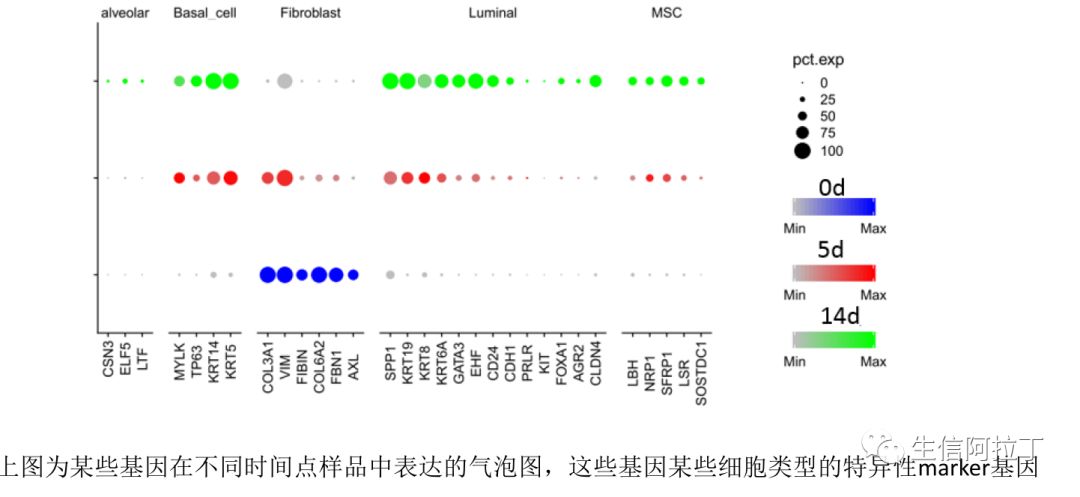
8
基 因 集 富 集 分 析
除了对常规的差异基因或者marker基因的功能分析以外,还有一种就是对某个基因集进行富集分析或者说打分。这种分析方式不需要在不同样品之间进行比较,关注点是某个基因集在每个细胞、每个细胞亚群中的富集程度,其中这样分析的内容可就多了。
基因集可以根据需要进行设置,比如关注T细胞激活的话,给定T细胞激活的基因集,就可以看到每个细胞的T细胞激活程度。基因集分析比较常用的工具是GSVA(Gene Set Variation Analysis),链接如下:
http://www.bioconductor.org/packages/release/bioc/html/GSVA.html
这里需要注意的是该工具尽量不要使用counts表达矩阵作为输入数据,如果实在需要用counts表达矩阵作为输入文件的话,需要修改GSVA的参数kcdf,设为Poisson,其默认为Gaussian。但是使用Poisson的时候,其耗时巨大,因此不建议使用counts表达矩阵。
一般建议使用连续性值,比如log-CPMs, log-RPKMs or log-TPMs,这样分析速度快很多,一般两三万细胞的话,30个基因集以内的,24小时内就可以完成。
至于基因集的设置,一般可以使用GO Term(去东方,最好用的在线GO富集分析工具)、KEGG代谢通路(20W+喜爱的Pathview网页版 | 整合表达谱数据KEGG通路可视化)、reactome代谢通路(没钱买KEGG怎么办?REACTOME开源通路更强大)、或者GSEA官方基因集(一文掌握GSEA,超详细教程)、自己提供的基因集都可以,下图为GSVA结果展示。
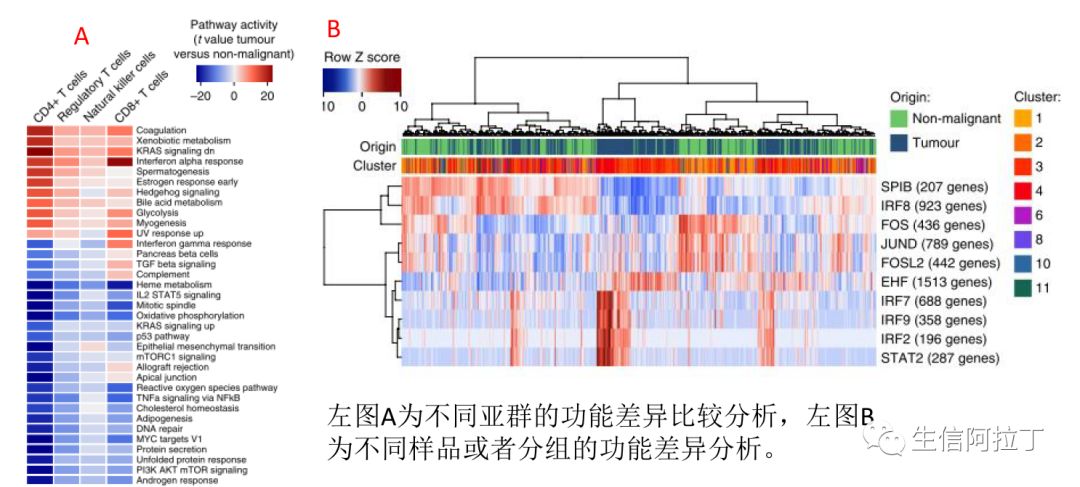
9
单细胞转录组发育轨迹分析
发育轨迹分析即拟时间分析,就是根据细胞中基因的表达量,基于特定基因对细胞进行排序的一个过程,其结果主要反映细胞发育的先后。
一般软件得到的结果是没法确定发育起始点,需要根据某些基因来判断发育起始点,这里特定基因可以是软件自动计算差异的基因,也可以是已知的跟发育相关的基因。其中最常用的发育轨迹的工具是monocle。
http://cole-trapnell-lab.github.io/monocle-release/
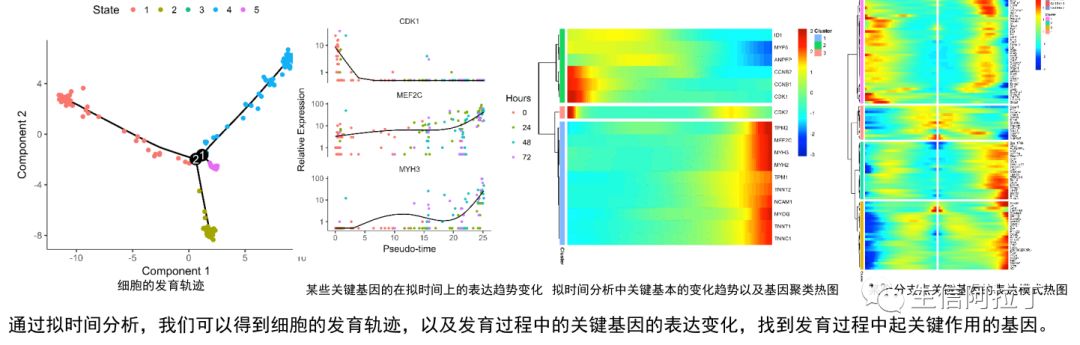
当然发育轨迹分析的工具有很多(NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集),目前能做此类分析结果的工具至少有60+以上,比如dyno (https://github.com/dynverse/dynmethods)工具搜集了60+,做了一个所有拟时间分析的集合,包括常用的monocle (http://cole-trapnell-lab.github.io/monocle-release/)、PAGA (https://github.com/theislab/paga)、pCreode (https://github.com/KenLauLab/pCreode)等等,不过此软件是基于docker,因此需要系统有root权限。
不同的单细胞转录组数据具有不同的特征,可能某个软件并不通用的。因此dyno工具通过其前期的表达特征,提供该数据对所有软件的最优适配的方案,可以选择最优的方案。
不过此工具也有局限性,比如工具不全、工具的版本更新不及时(monocle现在为版本2)、某些功能的缺失、与其他工具的兼容性有待提升等等。因此没有任何万能的工具,只能根据需要进行挑选工具。
参考文献
1.Giovanni Iacono, Ramon Massoni-Badosa, Holger Heyn. Single-cell transcriptomics unveils gene regulatory network plasticity[J]. Genome biology, 2019, 20(1).
2.Gioele L M , Ruslan S , Amit Z , et al. RNA velocity of single cells[J]. Nature, 2018.
3.Park J , Shrestha R , Qiu C , et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease[J]. Science, 2018:eaar2131.
4.Zhang X, Lan Y, Xu J, et al. CellMarker: a manually curated resource of cell markers in human and mouse[J]. Nucleic Acids Research, 2019.
5.Aran D, Looney A P, Liu L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage[J]. Nature Immunology, 2019, 20(2): 163-172.
6.Aibar S , González-Blas, Carmen Bravo, Moerman T , et al. SCENIC: single-cell regulatory network inference and clustering[J]. Nature Methods, 2017.
7.Wouter, Saelens, Robrecht, et al. A comparison of single-cell trajectory inference methods[J]. Nature Biotechnology, 2019.
8.F, Alexander, Wolf, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells.[J]. Genome biology, 2019.
9.Diether L , Els W , Bram B , et al. Phenotype molding of stromal cells in the lung tumor microenvironment[J]. Nature Medicine, 2018.
10.Zheng C , Zheng L , Yoo J K , et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing[J]. Cell, 2017, 169(7):1342-1356.e16.














![[SQL] 数据库图形化安装和使用](https://i-blog.csdnimg.cn/direct/68a832f9c2954f1a97f011996895b446.png)



