PIDiff 是一个针对蛋白质口袋特异性的、物理感知扩散的 3D 分子生成模型,通过考虑蛋白质-配体结合的物理化学原理来生成分子,在原理上,生成的分子可以实现蛋白-小分子的自由能最小。
一、背景介绍
PIDiff 来源于延世大学计算机科学系的 Sanghyun Park 教授为通讯作者的文章:《PIDiff:Physics informed diffusion model for protein pocket-specific 3D molecular generation》。文章链接:https://www.sciencedirect.com/science/article/pii/S0010482524009508。该文章于 2024 年 7 月 7 日发表在 Computers in Biology and Medicine 上。

在药物开发中,设计能够与目标蛋白质结合的药物是至关重要的。然而,大多数现有方法主要集中于在 3D 空间中对配体的几何信息进行建模,未能考虑蛋白质和配体的结合是由内在的物理化学原理驱动的现象。基于这种理解,作者提出了 PIDiff ,通过考虑蛋白质-配体结合的物理化学原理来生成分子的模型。PIDiff 不仅可以学习复合物的结构信息,还能学习最小化蛋白质-配体结合自由能。PIDiff 还引入了一个评估模型的实验框架,涵盖了生成模型在实际药物开发中应用的各种基本面。PIDiff 在 CrossDocked 2020 基准数据集上的表现优于基准模型,展示了其优越性。多种实验结果展示了 PIDiff 在实际药物开发中的潜在前景。
二、模型介绍
当前基于结构的分子生成模型存在两个主要限制:(1)现有方法仅学习分子的几何形状,并没有考虑分子结合到蛋白质口袋中的物理化学原理,忽略了分子相互作用和结合过程的深层次方面。(2)当前使用的蛋白质-配体数据集把结构表示为固定的三维坐标,而这些结构在实际环境中是处于动态变化的。现有的生成模型主要依赖这些固定三维坐标数据的经验分布进行训练,在生成准确反应分子相互作用真实本质的高质量分子面临挑战。
为解决这两个当前方法的限制,作者从物理感知神经网络(PINN, physics-informed neural network)中获得灵感,提出在生成模型中加入内在的物理化学原理。PINN是一个通过在神经网络中整合相关物理属性作为归纳偏差的综合框架,其根植于广泛的流体动力学研究中。通过利用这一概念,作者开发了 PIDiff,它基于蛋白质和配体结合自由能最小化的基本物理化学原理定义归纳偏差。PIDiff 不仅旨在学习结合到目标蛋白质的分子的几何信息,还被训练以确保生成的分子与目标蛋白质的结合自由能最小化。结合自由能可以近似为分子间相互作用能的总和,通过对生成原子的位置信息进行微分,作者在生成模型中引入结合自由能最小化条件。PIDiff 不仅学习了现有训练数据集的经验分布,还确保符合数据集中隐含的物理定律,从而允许生成更接近现实场景的高质量数据,比以前只专注于几何信息能更准确地反映现实环境。
评估结果显示,PIDiff 达到了最先进的性能。除了在测试集上评估结合亲和力、合成可及性和物理化学有效性等基准性能外,作者还验证了 PIDiff 模型在从不同的口袋结构中生成高结合亲和力分子的能力。作者还通过验证脱靶效应的选择性进行了额外评估。最后,作者通过整合分子动力学模拟(MD)来更现实地评估模型生成的分子在蛋白质口袋内的实际行为,完成了作者提出的全面评估框架。
总的来说,本研究的关键贡献是:
(1)作者提出了 PIDiff,这是 第一个结合了蛋白质和配体在结合自由能最小化状态下的 3D 分子生成模型。PIDiff 通过满足可应用于现实世界的物理定律,使生成的分子生成更符合真实世界。
(2)作者的实验结果表明,PIDiff 在 CrossDocked 2020 基准数据集上生成的分子对蛋白质口袋的结合亲和力方面,超过了 AR、liGAN、GraphBP、Pocket2Mol、DiffSBDD、TargetDiff 和 ResGen 等基准模型,创下了最先进的性能记录。
(3)作者提出了一个多样化和严格的实验框架,从生成模型在 SBDD 中的实用性角度评估模型的性能。性能评估的结果提供了分子生成模型在药物开发中应用的各种视角。
2.1 模型框架
如下图所示,作者提出了一种基于扩散的生成模型 PIDiff,该模型融合了蛋白质与配体之间的稳定状态的化学原理。PIDiff 通过固定蛋白质原子的位置,并在真实分子的原子位置和类型上注入噪声,经过一个前向过程将其转变为噪声分子。然后,模型通过一个逆向过程去除噪声,恢复回真实分子。模型的一个关键方面是在逆向过程中,不仅仅是重建真实分子原子的位置和类型,而是设计确保配体满足与目标蛋白质稳定结合的物理化学原理。在逆向过程中注入专业知识,以确保推断出(生成)的真实分子与蛋白质口袋之间的反应坐标处的结合自由能被最小化。

在传统的 3D 分子生成扩散模型中,训练的目标为在各个时间步下原子坐标 KL 散度损失,以及节点类型的后验 KL 散度损失,对应文章中:

这一部分的损失使得扩散模型生成的数据分布与真实分布之间对齐。
从物理学视角,需要基于扩散模型生成的原子坐标推测出蛋白和小分子之间的结合自由能。作者使用 Lennard-Jones 势能,计算蛋白和小分子之间的结合自由能。最小化结合自由能对于实现蛋白质和配体之间稳定且良好的结合至关重要。为此,作者将最小化自由能作为优化过程中的首要目标。引入了约束,以确保结合自由能对推断的原子位置的导数为零,让生成的分子与蛋白之间形成稳定的相互作用。
总 Lennard-Jones 势能表示为所有可能的蛋白质原子和配体原子对的能量总和,对应作者的如下公式:


因此,PIDiff 的损失除了节点和坐标的 KL 散度损失,还有 LJ 势能梯度的损失(让蛋白-小分子的 LJ 势能导数为0,那么蛋白和小分子的结合状态的能量处于凹点,即稳态)。
因为 LJ 势能的梯度损失添加,因此,PIDiff 模型需要重新训练,不能像TagMol 一样继承 TargetDiff 的checkpoint。
2.2 数据集和评价指标
为了验证训练好的生成模型是否能够生成可以与蛋白质口袋结合的分子,作者采用了CrossDocked 2020 数据集。作者对数据集进行了过滤和拆分,测试集和训练集是在蛋白质序列相似性低于40%的条件下划分。数据处理后,作者获得了一个包含 100,000 个蛋白质-配体对的高质量训练集和一个由 100 个蛋白质组成的测试集。
作者通过结合亲和力,物理化学有效性等指标评价生成分子。VinaScore 直接根据生成的 3D 分子估算结合亲和力;VinaMin 在估算前执行局部结构最小化;VinaDock 涉及额外的重新对接过程,反映出最佳可能的结合亲和力;HighAffinity 则衡量生成的分子中有多少在每个测试蛋白质中比参考分子具有更好的结合能力。 表示合成可及性(SA)小于 6 的生成分子的 VinaScore ,综合考虑了结合亲和力和合成性。SR(Success Rate)定义为测试集中100 个蛋白质口袋中至少有一个生成分子表现出比测试集中口袋中的配体更强结合亲和力的比例。之后通过 PoseBusters 评估生成分子的物理化学有效性,通过评估的分子被定义为有效分子,生成分子中有效分子的比例记作
。
2.3 模型性能
有效的模型应该被用来提高效率并加速药物发现过程,为了验证 PIDiff 能否实际应用于药物发现,作者通过回答下面五个问题来进行探究:
- 由模型生成的分子能有效结合到目标蛋白质上吗?
- 由模型生成的分子在结合目标蛋白质时能保持稳定状态吗?
- 由模型生成的分子是否能够有效结合到针对实际疾病的蛋白质上,根据各种来源的结构?
- 由模型生成的分子针对特定蛋白质结构是否能对具有不同结构的蛋白质表现出选择性?
- 由模型生成的分子在与真实药物比较时,能在实际生物系统中保持稳定的结合吗?
2.3.1 生成分子与目标蛋白质结合亲和力的分析
如下表所示,PIDiff 在六个评估指标中大大优于其他模型。与之前的最先进模型 TargetDiff 相比,PIDiff 在 VinaScore 上提高了 21%,在 HighAffinity 上提高了 12%,在 上提高了 13%,在 SR 上提高了 9% 。如 SR 指标所示,PIDiff 可以生成在测试集中的所有蛋白质口袋中比参考分子表现出更强结合的分子。这些结果表明,先验的化学知识(即蛋白质和配体的复杂结构应位于结合自由能景观的最小值)对合理的分子设计至关重要。这在将PIDiff 与TargetDiff 进行比较时尤其明显,后者也使用扩散生成模型来生成分子。

此外,PIDiff 模型的消融情况不考虑物理化学原理时,表现相当于 TargetDiff 的表现。在和 PIDiff 作比较的基准模型的生成分子中,会出现正值的结合能(VinaScore),这对应不合理的结合姿势,但重新对接会修正不合理的构象,VinaDock 会变成正值。所以判断生成分子的构象合理性,应着重查看 VinaScore 。
在 指标方面,PIDiff 的性能排名第三,仅次于 Pocket2Mol 和 AR ,主要是由于许多分子未能通过键角测试。对于 Pocket2Mol 和 AR 等模型,分子生成过程采用自回归采样方法,其中原子按顺序预测以形成完整的分子。这一过程确保在预测下一个原子时保持先前预测原子之间的几何合理性。因此,许多分子在 PoseBusters 套件中的键角测试中通过。然而,PIDiff 采用非自回归采样方法,推断整个分子而不是按顺序预测原子。因此,它在键角测试中的表现相对低于自回归采样模型。然而,自回归采样模型由于训练和采样过程之间的差异,容易出现累积错误和暴露偏差。除
指标外,这些模型在所有评估指标中的表现均显著低于PIDiff 。这表明这些模型未能生成对目标蛋白质具有强结合亲和力的分子,使其不适合作为药物候选者。因此,自回归模型并不适合基于结构的药物设计中的生成任务。
2.3.2 生成分子在结合能方面的稳定性
作者通过比较生成分子在对接前后构象的 RMSD 来验证生成分子的稳定性。下图展示了各模型生成的分子在对接前后构象差异的分布。与TargetDiff, Pocket2Mol, ResGen 五个比较模型生成的分子相比,PIDiff 模型生成的分子在对接后表现出显著较少的 RMSD 变化。这一结果清楚地表明,PIDiff 能够推断出最有利于结合的分子。此外,这也突显了在 PIDiff 模型训练期间不仅要学习分子位置和结构的分布,还要在生成模型中融入先前的物理化学知识的重要性。

下图用两个示例说明对接前后差异的两种情况。案例 A 展示了生成分子对接前后构象差异较大的情况。这意味着在对接过程中,对生成分子进行了显著的构象调整,以实现理想的蛋白质-配体结合。因此,这表明生成的分子在与蛋白质结合时,其结构和姿态不利于结合。案例 B 展示了生成分子对接前后构象差异较小的情况,表明生成的分子在与蛋白质结合时,其结构和姿态具有优势。

以下是几个案例。在下图中,对比了口袋中的原始配体分子和 PIDiff 、ResGen 生成的分子。青色表示对接前的分子,红色表示对接后的分子。由 PIDiff 模型生成的分子在对接前后表现出微不足道的构象差异。相比之下,由 ResGen 生成的分子在对接后显示出显著的构象变化,表明这些分子需要大量微调才能与蛋白质进行有利结合,也符合 RMSD 值的分布结果。

2.3.3 生成分子对各种疾病靶蛋白的适用性
为了评估3D分子生成的实际效用,验证这些模型是否能为实际药物靶标蛋白设计合理的分子,作者选择了几个具有代表性的激酶靶点,包括 RET、ERBB2、ABL1、ALK、EGFR 和 KIT 。下图展示了生成分子的结合亲和力分布和参考药物的比较。PIDiff 生成的分子中,具有比参考药物更高结合亲和力的分子比例为:RET 为 36%,ERBB2 为 13%,ABL1 为 38%,EGFR 为 84%,ALK 为 35%,KIT 为 52% 。值得注意的是,为 EGFR 蛋白生成的大多数分子表现出比结合药物更高的结合亲和力。此结果说明,PIDiff 不仅在基准数据集上取得了高性能,而且还具备在实际中生成具有竞争性结合亲和力的靶蛋白分子的能力。

当前发现的大部分蛋白质只知道序列信息,并没有结构信息。为了评估 PIDiff 在没有结构的蛋白上的表现,作者选择了 FLT3 和 VEGFR2 蛋白,分别根据序列预测结构和从 PDB 数据库获取结构(FLT3: 4RT7, VEGFR2: 1YWN)生成 1000 个分子并计算 Vina Score。下图是生成分子和参考药物(Sunitinib)的 Vina Score 比较。AlphaFold 蛋白生成的 1000 个分子的 Vina Score 分布显示为灰色,而基于 PDB 蛋白生成的 1000 个分子的 Vina Score 分布显示为绿色。参考药物的 Vina Score 以蓝色条表示。在 FLT3 和 VEGFR2 两个蛋白上,AlphaFold 蛋白生成的分子中比参考药物结合亲和力更高的比例分别达到 58% 和 74% 。并且基于 AlphaFold 蛋白和 PDB 蛋白的 Vina Score 分布并没有显著差异。这表明 PIDiff 有能力使用从 AlphaFold 获得的结构为靶标蛋白生成具有竞争力的分子。

2.3.4 生成分子的选择性
生成分子的选择性指的是基于特定蛋白质结构生成的分子对具有不同结构的蛋白质的结合相对较弱。为了验证 PIDiff 生成分子的选择性,作者选择了 B-Raf 蛋白和其抑制剂(sorafenib),并确定 PKB、ERK1、PIM1、MEK1、CDK1、PKC、IGFR1、EGFR、c-MET、HER2 为脱靶蛋白进行对比。基于 B-Raf 蛋白生成分子后,选定 SA 小于 6 的分子,计算这些选定分子和这些蛋白的 Vina Score 。下图是选定分子在靶蛋白和脱靶蛋白上的 Vina Score 分布,可以看出 PIDiff 为 B-Raf 生成的分子往往对结构不同于靶蛋白的脱靶蛋白结合力较弱,这表明了 PIDiff 在 SBDD 中的潜在应用,表明了 PIDiff 在实现高度选择性靶向结合方面分子生成的实用性。

2.3.5 从热力学角度看生成分子的稳定性
为了评估生成分子的稳定性,作者选择 2.3.4 中 B-Raf 蛋白生成的分子,根据 ECFP 指纹进行 k-means 聚类(k = 3),从每个聚类中选择一个分子使用 ABMER 在10 ns上进行 MD 模拟。同时也对 B-Raf 蛋白的抑制剂(sorafenib)进行 MD 模拟。如下图所示,与抑制剂随时间的RMSD 相比,PIDiff 生成的三个分子结构随时间表现出稳定的波动范围,整体上显示了较低的RMSD 曲线。因此可以得出结论,生成分子可以稳定结合到靶标蛋白上,即使与已批准的抑制剂相比,也具有竞争性的结合能力。

三、PIDiff 评测
3.1 安装环境
复制代码项目:
git clone https://github.com/hello-maker/PIDiff.git创建 PIDiff 环境
conda create -n PIDiff python=3.8
conda activate PIDiff
conda install pytorch=1.13.1 pytorch-cuda=11.6 -c pytorch -c nvidia
conda install pyg=2.2.0 -c pyg
conda install rdkit=2022.03.2 openbabel=3.1.1 tensorboard=2.13.0 pyyaml=6.0 easydict=1.9 python-lmdb=1.4.1 -c conda-forge
# 安装 vina 对接所需的依赖库
pip install meeko==0.1.dev3 scipy pdb2pqr vina==1.2.2
python -m pip install git+https://github.com/Valdes-Tresanco-MS/AutoDockTools_py33.2 训练模型
3.2.1 下载项目数据
作者把项目用到的数据保存在谷歌网盘,链接是 https://drive.google.com/drive/folders/1qzuYX39_apCWcZ6yFMkY9RYAI8ijfmvY 。

其中 Training Data 文件夹中是预处理好的 CrossDock 数据集。test_set 文件夹中是测试数据,包含 100 个蛋白和配体。创建 ./data 文件夹,把 Training Data 文件夹中的文件放在 ./data 文件夹中,同时把 test_set 的文件也放在 ./data 文件夹中
完整的 PIDiff 项目目录如下:
.
|-- README.md
|-- assets
|-- configs
|-- data
|-- datasets
|-- logs_diffusion
|-- models
|-- nohup.out
|-- sampling_results
|-- scripts
|-- test_sampling.sh
`-- utils
9 directories, 3 files其中,./data 文件夹的目录如下:
.
|-- crossdocked_pocket10_pose_split.pt
|-- crossdocked_v1.1_rmsd1.0_pocket10_processed_final.lmdb
|-- index.pkl
|-- test.log
`-- test_set
1 directory, 4 files此外,由于 PIDiff 的脚本中使用的是绝对路径,所以项目下载之后需要修改路径。为方便后续训练和评测,我们把项目中的绝对路径进行修改。
(1)scripts/train_diffusion.py 中第 4 行的
sys.path.append("/home/csy/work/3D/PIDiff")和 39 行的
os.chdir('/home/csy/work/3D/PIDiff')和 49 行的
args.config = '/home/csy/work/3D/PIDiff/configs/training.yml'分别修改为
sys.path.append("../PIDiff")和
os.chdir('../PIDiff')和
args.config = './configs/training.yml'(2)configs/sampling.yml 中第 2 行的
checkpoint: /home/csy/work/3D/PIDiff/logs_diffusion/修改为
checkpoint: ./logs_diffusion/(3)scripts/evaluate_diffusion.py 中第 3 行的
sys.path.append("/home/csy/work/3D/PIDiff")修改为
sys.path.append("../PIDiff")(4)scripts/sample_diffusion.py 中第 5 行的
sys.path.append("/home/csy/work/3D/PIDiff")修改为
sys.path.append("../PIDiff")(5)models/architecture.py 中第 11 行的
sys.path.append("/home/csy/work/3D/PIDiff")修改为
sys.path.append("../PIDiff")3.2.3 配置训练参数及处理报错
项目没有提供训练好的模型,需要我们训练模型,模型训练的默认配置文件在 ./configs/training.yml,最大迭代次数(max_iters)设置为 1000000
训练模型的命令如下:
python scripts/train_diffusion.py 报错如下:
Traceback (most recent call last):
File "scripts/train_diffusion.py", line 13, in <module>
import wandb
ModuleNotFoundError: No module named 'wandb'缺少 wandb 模块,通过 conda 安装:
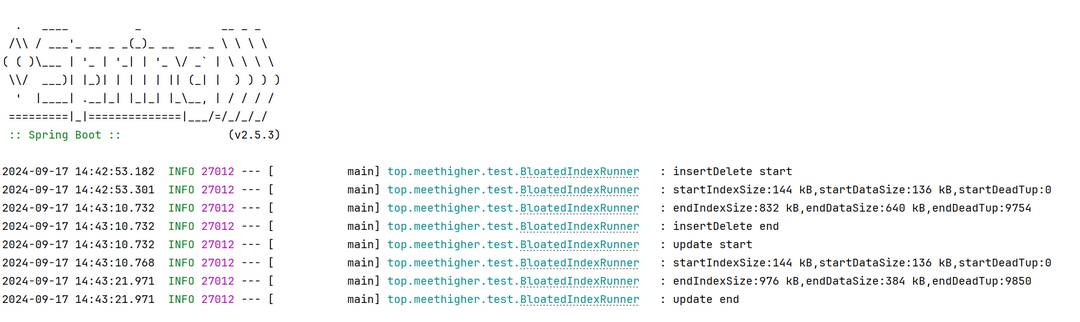
conda install wandb安装 wandb 之后,训练模型的命令可以正常执行。训练好的模型保存在 ./logs_diffusion/training_2024_08_28__11_14_11/checkpoints 中,每个100次迭代保存一个 pt 文件。
3.3 分子生成案例测试
我们使用自己训练好的模型进行分子生成测试,修改 ./configs/sampling.yml ,指定训练好的模型,具体参数设置如下,默认生成 100 个分子。
model:
checkpoint: ./logs_diffusion/training_2024_08_28__11_14_11/checkpoints/30600.pt
sample:
seed: 2021
num_samples: 100
num_steps: 1000
pos_only: False
center_pos_mode: protein
sample_num_atoms: prior3.3.1 内置案例
测试集中 index 为 0 的体系(PDB : 2Z3H,对应的保存路径为:./data/test_set/BSD_ASPTE_1_130_0 )的口袋以及原来小分子的展示如下图,我们把这个蛋白作为测试分子生成的内置案例。

分子采样的脚本是 ./scripts/sample_diffusion.py,使用编号为 0 的蛋白进行分子生成,命令如下:
python scripts/sample_diffusion.py \
configs/sampling.yml \
--data_id 0 \
--result_path ./result_0 --data_id 0 指定分子生成的编号,这里指定编号为 0 的蛋白,--result_path ./result_0 指定生成分子保存到 ./result_0 中。batch_size 设置为 16 显存为 20 G, 这个体系生成 100 个分子大约花费半个小时。
命令报错:
Traceback (most recent call last):
File "scripts/sample_diffusion.py", line 17, in <module>
from models.architecture import PIDiff, log_sample_categorical
File "/workspace/GuanXL/projects/PIDiff/models/architecture.py", line 8, in <module>
from models.egnn import EGNN
ModuleNotFoundError: No module named 'models.egnn'报错解决方法,详见完整版文档,这里不累述。
解决报错后,采样命令可以正常运行,整个采样过程约半个小时,运行结果如下:
Successfully load the dataset (size: 100)!
Successfully load the model! ./logs_diffusion/training_2024_08_28__11_14_11/checkpoints/30600.pt
100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1/1 [23:22<00:00, 1402.74s/it]
Sample done!采样结果保存在指定的输出文件夹 ./result_0,内容如下:
.
|-- result_0.pt
`-- sample.yml
0 directories, 2 files接着评估生成的分子,命令如下:
python scripts/evaluate_diffusion.py \
./result_0 \
--docking_mode vina_score\
--protein_root ./data/test_set注:当 --docking_mode qvina 或者 --docking_mode none 是可以正常完成评估的。但是 --docking_mode vina_score 或者 --docking_mode vina_dock 就会出现错误,此处已修改代码。
此外,在 PIDiff 使用 scripts/evaluate_diffusion.py 脚本生成和评价分子的过程中,并没有保存生成的分子构象,所以我们修改评价分子的脚本 evaluate_diffusion.py,加入保存分子构象的代码。
评估分子的运行输出如下:
[2024-09-04 02:53:12,631::evaluate::INFO] Evaluate done! 100 samples in total.
[2024-09-04 02:53:12,632::evaluate::INFO] mol_stable: 0.0000
[2024-09-04 02:53:12,632::evaluate::INFO] atm_stable: 0.6263
[2024-09-04 02:53:12,632::evaluate::INFO] recon_success: 0.9900
[2024-09-04 02:53:12,632::evaluate::INFO] eval_success: 0.9100
[2024-09-04 02:53:12,632::evaluate::INFO] complete: 0.9100
[2024-09-04 02:53:12,635::evaluate::INFO] JS bond distances of complete mols:
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-6|4: 0.4721
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-6|1: 0.5220
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-8|1: 0.4984
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-7|1: 0.4488
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-8|2: 0.4962
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-6|2: 0.4745
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-7|4: 0.5537
[2024-09-04 02:53:12,635::evaluate::INFO] JSD_6-7|2: 0.5406
[2024-09-04 02:53:12,657::evaluate::INFO] JSD_CC_2A: 0.4520
[2024-09-04 02:53:12,658::evaluate::INFO] JSD_All_12A: 0.1467
[2024-09-04 02:53:12,658::evaluate::INFO] Atom type JS: 0.0935
[2024-09-04 02:53:12,818::evaluate::INFO] Number of reconstructed mols: 99, complete mols: 91, evaluated mols: 91
[2024-09-04 02:53:12,818::evaluate::INFO] QED: Mean: 0.263 Median: 0.225
[2024-09-04 02:53:12,818::evaluate::INFO] SA: Mean: 0.501 Median: 0.490
[2024-09-04 02:53:12,819::evaluate::INFO] VALID: Mean: 1.000
[2024-09-04 02:53:12,819::evaluate::INFO] Vina Score: Mean: -9.341 Median: -9.002
[2024-09-04 02:53:12,819::evaluate::INFO] Vina Min : Mean: -9.464 Median: -9.111
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 3 ratio: 0.000
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 4 ratio: 0.176
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 5 ratio: 0.407
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 6 ratio: 0.780
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 7 ratio: 0.429
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 8 ratio: 0.088
[2024-09-04 02:53:12,819::evaluate::INFO] ring size: 9 ratio: 0.033
Generated molecules saved as sdf format!从返回结果可以看出,能够重构的生成分子有 91 个,QED 和 SA 均值分别为 0.263 和 0.501,生成分子中包含 五、六、七环的比例分别为 0.407、0.780 和 0.429。评估结果和生成的分子构象默认保存在 ./result_0/eval_results 中。生成分子的 vina score 的均值为:-9.341。
所有生成的分子如下:
PIDiff_2z3h_outputs
vina score 排名前 3 的分子的 2D 结构如下,对应的 vina score 分数分别为 -14.184, -13.630, -13.595:

上述vina score 排名前 3 的分子在口袋中的 Pose 如下:



3.3.2 自定义案例
针对特定的分子生成,作者提供了一个Ipynb文件(./scripts/real_world/Inference.ipynb)。下面根据提供的 Ipynb 进行测试。
导入相关的模块:
import os
import sys
os.chdir('/home/csy/work/3D/PIDiff')
sys.path.append("/home/csy/work/3D/PIDiff")
sys.path.append("/home/csy/work/3D/PIDiff/utils")
sys.path.append("/home/csy/work/3D")
import argparse
import multiprocessing as mp
import pickle
import shutil
from functools import partial
from tqdm.auto import tqdm
from utils.data import PDBProtein, parse_sdf_file
from mol_tree import *
import rdkit
import rdkit.Chem as Chem
import rdkit.Chem.AllChem as AllChem
from openbabel import pybel
from openbabel import openbabel as ob
import subprocess
import os
import numpy as np
import shutil
from plip.structure.preparation import PDBComplex
import traceback
import utils.misc as misc
import utils.transforms as trans
from torch_geometric.transforms import Compose
from models.architecture import PIDiff, log_sample_categorical
from scripts.sample_diffusion import sample_diffusion_ligand导入模块报错,缺少 mol_tree 模块:
ModuleNotFoundError Traceback (most recent call last)
Cell In[2], line 8
6 from tqdm.auto import tqdm
7 from utils.data import PDBProtein, parse_sdf_file
----> 8 from mol_tree import *
9 import rdkit
10 import rdkit.Chem as Chem
ModuleNotFoundError: No module named 'mol_tree'解决办法,去 JunctionTreeVAE 项目中复制过来即可。
定义蛋白和小分子结构预处理函数:
def sdf2pdb(sdf_path, pocket_path):
output_path = sdf_path.replace('.sdf', '.pdb')
run_line = f'obabel -isdf {sdf_path} -opdb -O {output_path}'
result = subprocess.run(run_line.split(), capture_output=True, check=True, universal_newlines=True)
print(result)
complex_path = pocket_path.replace('.pdb', 'complex.pdb')
shutil.copy(pocket_path, complex_path)
comp = open(complex_path, 'a')
comp.write("TER\n")
lig_pdb = open(output_path)
lines = lig_pdb.readlines()
for line in lines:
if line[0:6] == 'HETATM' or line[0:6] == 'CONECT':
comp.write(line)
#comp.write("END")
lig_pdb.close()
comp.close()
return complex_path
def load_item(sdf_path, pdb_path):
with open(pdb_path, 'r') as f:
pdb_block = f.read()
with open(sdf_path, 'r') as f:
sdf_block = f.read()
return pdb_block, sdf_block
def process_item(sdf_path, pdb_path, pocket_path):
pdb_block, sdf_block = load_item(sdf_path, pdb_path)
protein = PDBProtein(pdb_block)
# ligand = parse_sdf_block(sdf_block)
ligand = parse_sdf_file(sdf_path)
pdb_block_pocket = protein.residues_to_pdb_block(
protein.query_residues_ligand(ligand, 20)
).replace("END\n", "")
pocket_path = pocket_path + pdb_path.split('/')[-1].replace('.pdb', '_pocket.pdb')
with open(pocket_path, 'w') as f:
f.write(pdb_block_pocket)
return pocket_path下载 PDB ID 为 3WZE 的蛋白作为测试案例,使用 pymol 把配体保存为 ligand.sdf,去除溶剂,离子等只保留蛋白结构,保存为 receptor.pdb。3WZE 口袋与原来配体小分子的如下图:

在 ./scripts/real_world 文件夹中创建 3wze 文件夹,文件结构如下:
.
|-- 3wze
|-- Inference.ipynb
`-- MD.ipynb
1 directory, 2 files其中,3wze 文件夹包括处理好的蛋白结构和配体结构,具体如下:
.
|-- ligand.sdf
`-- receptor.pdb
0 directories, 2 files指定配体和蛋白的路径:
protein_name = '3wze'
sdf_path = f'./scripts/real_world/{protein_name}/ligand.sdf'
pdb_path = f'./scripts/real_world/{protein_name}/receptor.pdb'转换并处理蛋白质和配体结构:
pocket_path = f'./scripts/real_world/{protein_name}/'
pocket_path = process_item(sdf_path, pdb_path, pocket_path)
complex_path = sdf2pdb(sdf_path, pocket_path)
pocket_path = f'./scripts/real_world/{protein_name}/receptor_pocket.pdb'
complex_path = f'./scripts/real_world/{protein_name}/receptor_pocketcomplex.pdb'
得到蛋白质口袋结构,小分子的 PDB 结构以及复合物结构,处理后的结构保存在 ./scripts/real_world/3wze 中,文件结构如下:
.
|-- ligand.pdb
|-- ligand.sdf
|-- receptor.pdb
|-- receptor_pocket.pdb
`-- receptor_pocketcomplex.pdb
0 directories, 5 files定义数据处理的类 ProteinLigandData
import torch
import torch_scatter
import numpy as np
from torch_geometric.data import Data
from torch_geometric.loader import DataLoader
class ProteinLigandData(Data):
def __init__(self, *args, **kwargs):
super().__init__(*args, **kwargs)
@staticmethod
def from_protein_ligand_dicts(protein_dict=None, ligand_dict=None, residue_dict=None, inter_dict=None, **kwargs):
instance = ProteinLigandData(**kwargs)
if protein_dict is not None:
for key, item in protein_dict.items():
instance['protein_' + key] = item
if ligand_dict is not None:
for key, item in ligand_dict.items():
instance['ligand_' + key] = item
if residue_dict is not None:
for key, item in residue_dict.items():
instance[key] = item
if inter_dict is not None:
for key, item in inter_dict.items():
instance[key] = item
instance['ligand_nbh_list'] = {i.item(): [j.item() for k, j in enumerate(instance.ligand_bond_index[1])
if instance.ligand_bond_index[0, k].item() == i]
for i in instance.ligand_bond_index[0]}
"""
Here it for "residue_pos"
"""
instance['residue_pos'] = torch.stack([instance['pos_CA'],
instance['pos_C'],
instance['pos_N'],
instance['pos_O']], dim=1)
return instance
def __inc__(self, key, value, *args, **kwargs):
if key == 'ligand_bond_index':
return self['ligand_element'].size(0)
else:
return super().__inc__(key, value)
def torchify_dict(data):
output = {}
for k, v in data.items():
if isinstance(v, np.ndarray):
output[k] = torch.from_numpy(v)
else:
output[k] = v
return output
处理原始的结构数据,获取相互作用关系等作为模型的输入信息:
我们将 interactions = get_interaction(complex_path, sdf_path) 这行代码注释掉,运行下面的代码,代码修改成:
# interactions = get_interaction(complex_path, sdf_path)
pdb_block, sdf_block = load_item(sdf_path, pocket_path)
pocket_dict = PDBProtein(pdb_block).to_dict_atom()
residue_dict = PDBProtein(pdb_block).to_dict_residue()
ligand_dict = parse_sdf_file(sdf_path)
data = ProteinLigandData.from_protein_ligand_dicts(
protein_dict=torchify_dict(pocket_dict),
ligand_dict=torchify_dict(ligand_dict),
residue_dict=torchify_dict(residue_dict),
)
data.protein_filename = pdb_path
data.ligand_filename = sdf_path
#data = data.to_dict()在 GPU 上对 3wze 的口袋进行分子采样,配置文件和内置方案的相同,加载训练好的模型。
device = 'cuda:0'
# config_path = '/home/csy/work/3D/PIDiff/scripts/real_world_validation/sampling.yml'
config_path = './configs/sampling.yml' # wufeil
sample_size = 16 # 太大会爆内存
config = misc.load_config(config_path)
misc.seed_all(config.sample.seed)
ckpt = torch.load(config.model.checkpoint, map_location=device)
protein_featurizer = trans.FeaturizeProteinAtom()
ligand_atom_mode = ckpt['config'].data.transform.ligand_atom_mode
ligand_featurizer = trans.FeaturizeLigandAtom(ligand_atom_mode)
transform = Compose([
protein_featurizer,
ligand_featurizer,
trans.FeaturizeLigandBond(),
])
model = PIDiff(
ckpt['config'].model,
protein_atom_feature_dim=protein_featurizer.feature_dim,
ligand_atom_feature_dim=ligand_featurizer.feature_dim
).to(device)
model.load_state_dict(ckpt['model'])
data = transform(data)进行分子生成
all_pred_pos, all_pred_v, pred_pos_traj, pred_v_traj, pred_v0_traj, pred_vt_traj, time_list = sample_diffusion_ligand(
model, data,
config.sample.num_samples,
batch_size=sample_size, device=device,
num_steps=config.sample.num_steps,
pos_only=config.sample.pos_only,
center_pos_mode=config.sample.center_pos_mode,
sample_num_atoms=config.sample.sample_num_atoms
)
batch_size 设置为 16 显存为 8 G, 在 3wze 的蛋白口袋中采样 100 个分子,用时约 1.5 个小时。把生成的分子信息保存到 result 中:
result = {
'data': data,
'pred_ligand_pos': all_pred_pos,
'pred_ligand_v': all_pred_v,
'pred_ligand_pos_traj': pred_pos_traj,
'pred_ligand_v_traj': pred_v_traj,
'time': time_list
}我们添加代码,把生成分子信息和采样的配置文件保存到 ./scripts/real_world/{result_path} 文件夹中,即./scripts/real_world/3wze 中。
# 保存分子生成结果
result_path = protein_name
os.makedirs(result_path, exist_ok=True)
shutil.copyfile(config_path, f'./scripts/real_world/{result_path}/sample.yml')
torch.save(result, f'./scripts/real_world/{result_path}/result_1.pt')
评价生成的分子,具体命令如下:
python scripts/evaluate_for_pocket.py \
./scripts/real_world/3wze/result_1.pt \
--verbose True \
--protein_path ./scripts/real_world/3wze/receptor.pdb \
--docking_mode qvina \
--exhaustiveness 16 \
--save True第一个参数指定生成的分子信息,即 ./scripts/real_world/3wze/result_1.pt。生成分子要对接到蛋白口袋中,--protein_path ./scripts/real_world/3wze/receptor.pdb 指定对接的蛋白。--docking_mode qvina 指定生成分子的对接使用 qvina 。
运行输出:
Eval: 0%| | 0/1 [00:00<?, ?it/s]Best affinity: -8.3
Best affinity: -7.9
Best affinity: -1.3
Best affinity: -11.0
Best affinity: -3.0
[14:03:01] Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] ERROR: Could not sanitize molecule ending on line 99
[14:03:01] ERROR: Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] ERROR: Could not sanitize molecule ending on line 227
[14:03:01] ERROR: Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] ERROR: Could not sanitize molecule ending on line 355
[14:03:01] ERROR: Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] Explicit valence for atom # 25 N, 4, is greater than permitted
[14:03:01] ERROR: Could not sanitize molecule ending on line 483
[14:03:01] ERROR: Explicit valence for atom # 25 N, 4, is greater than permitted
[2024-09-05 14:03:01,379::evaluate::WARNING] Evaluation failed for 0_6
Best affinity: -8.7
Best affinity: -9.9
... ...评价分子前会先根据分子采样的节点类型和坐标信息重构分子构象,但有些不合法的分子不能重构构象。可以重构构象的有效分子会对接到蛋白口袋中,通过 qvina 计算对接打分。
运行结束后,整体生成分子的评估结果如下:
[2024-09-05 14:19:03,239::evaluate::INFO] Evaluate done! 100 samples in total.
[2024-09-05 14:19:03,240::evaluate::INFO] mol_stable: 0.0000
[2024-09-05 14:19:03,240::evaluate::INFO] atm_stable: 0.7520
[2024-09-05 14:19:03,240::evaluate::INFO] recon_success: 0.9000
[2024-09-05 14:19:03,240::evaluate::INFO] eval_success: 0.5400
[2024-09-05 14:19:03,240::evaluate::INFO] complete: 0.5600
[2024-09-05 14:19:03,243::evaluate::INFO] JS bond distances of complete mols:
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-6|4: 0.4348
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-6|1: 0.5169
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-8|1: 0.5723
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-7|1: 0.4054
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-8|2: 0.5718
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-6|2: 0.3662
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-7|4: 0.5659
[2024-09-05 14:19:03,243::evaluate::INFO] JSD_6-7|2: 0.4815
[2024-09-05 14:19:03,257::evaluate::INFO] JSD_CC_2A: 0.3709
[2024-09-05 14:19:03,257::evaluate::INFO] JSD_All_12A: 0.1341
[2024-09-05 14:19:03,257::evaluate::INFO] Atom type JS: 0.1382
[2024-09-05 14:19:03,422::evaluate::INFO] Number of reconstructed mols: 90, complete mols: 56, evaluated mols: 54
[2024-09-05 14:19:03,422::evaluate::INFO] QED: Mean: 0.431 Median: 0.459
[2024-09-05 14:19:03,422::evaluate::INFO] SA: Mean: 0.501 Median: 0.505
[2024-09-05 14:19:03,422::evaluate::INFO] Vina: Mean: -5.450 Median: -8.000
[2024-09-05 14:19:03,422::evaluate::INFO] ring size: 3 ratio: 0.000
[2024-09-05 14:19:03,423::evaluate::INFO] ring size: 4 ratio: 0.167
[2024-09-05 14:19:03,423::evaluate::INFO] ring size: 5 ratio: 0.630
[2024-09-05 14:19:03,423::evaluate::INFO] ring size: 6 ratio: 0.889
[2024-09-05 14:19:03,423::evaluate::INFO] ring size: 7 ratio: 0.574
[2024-09-05 14:19:03,423::evaluate::INFO] ring size: 8 ratio: 0.204
[2024-09-05 14:19:03,423::evaluate::INFO] ring size: 9 ratio: 0.056
Generated molecules saved as sdf format in ./scripts/real_world/3wze/eval_results/Generated_molecules.sdf!从返回结果可以看出,能够重构构象并评估的生成分子有 54 个,QED 和 SA 均值分别为 0.431 和 0.501,生成分子中包含 五、六、七环的比例分别为 0.630、0.889 和 0.574。生成分子的 qvina score 的均值为:-5.450,中位数值为:-8.00 ,对比 TagMol,以及之前的 IPDiff 这一结果并不是很好。从生成分子的有效数来说,也不是很好。评估结果和生成的分子构象默认保存在 ./scripts/real_world/3wze/eval_results 中。
所有生成的分子如下:
PIDiff_3wze_outputs
qvina score 排名前 3 的分子的 2D 结构如下,对应的 qvina score 分数分别为 -11.0, -10.7, -10.6:

qvina score 排名前 3 的分子在口袋中的 Pose 如下:



3.3.3 生成结构的 redocking RMSD
从生成分子的 qvina 打分来看,PIDiff 生成分子的结合力没有之前介绍的 TagMol,IPDiff 等模型好,但是 PIDiff 模型的特点是,深度神经网络不但要学会去噪恢复真实的分子,还要应对 LJ 势能梯度的损失。LJ 势能梯度损失,会使得蛋白-生成分子复合物能量最低,对应着文章中 redocking 的 RMSD 很小。
为了考察 PIDiff 生成的分子与蛋白之间的稳定性,我们对生成的 qvina score 最好的 top 3 分子做了 redocking。生成分子 pose 与 redocking pose 的比较如下图:
qvina score -11.0, redocking score -7.99:

qvina score -10.7, redocking score -8.3:

qvina score -10.6, redocking score -7.93:

从上述结果来说,LJ势能添加到损失中还是有作用的,前两个分子 redocking 的pose 非常相似,说明生成分子时 PIDiff 确实考虑了蛋白和分子之间的相互作用,生成的分子与蛋白结合的 pose 是稳定的。(注:对接 redcoking 打分差距大,是因为 redocking 的工具不是 qvina)
四、总结
作者提出了 PIDiff ,该模型不仅考虑蛋白质-配体的结构信息,还考虑蛋白质和配体结合的内在原理的必要性。在基准数据集和典型靶标蛋白质上的评估结果证明了 PIDiff 的实际应用性, PIDiff 具有加速药物发现的巨大潜力。
虽然作者对生成分子的结合能力、选择性和稳定性等方面做了探究,但是要作为候选药物,还需要考虑 ADMET(吸收、分布、代谢、排泄和毒性)等药效或药物特性,但作者并没有深入探究这些方面。如果要考虑药效特性,潜在空间优化和引导采样技术将是可供考虑的解决方案。









![24-9-17-读书笔记(十八)-《契诃夫文集》(二)上([俄] 契诃夫 [译] 汝龙 )](https://i-blog.csdnimg.cn/direct/119d4e8f7a1440d7a0fa84ddbf230581.png)