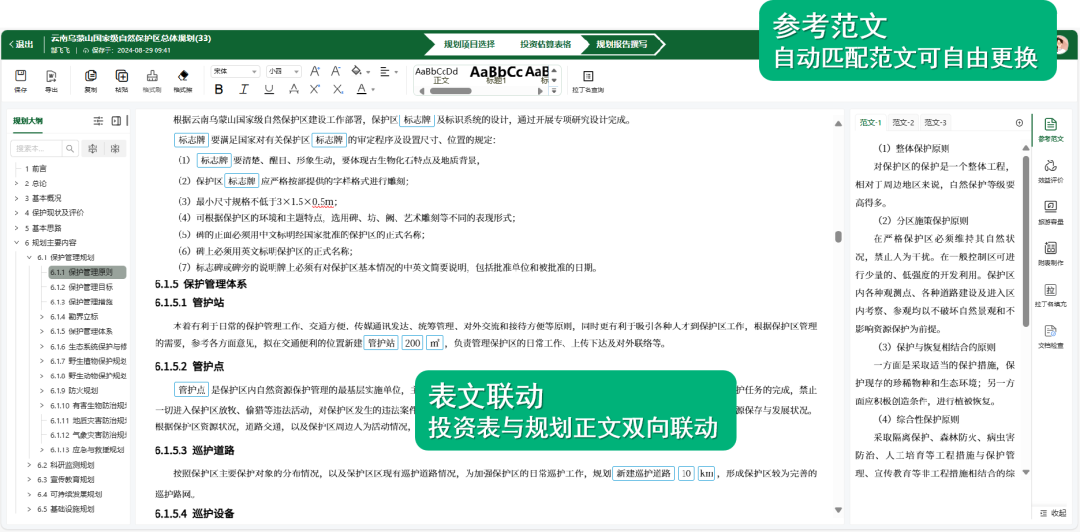
1、报错信息
The sequence "chr1" not defined in the header: chr1.recode.vcf
(Quick workaround: index the file.)

所使用的命令,目的是想合并所提取的特定染色体。
bcftools concat -O v /
-o varscan.indel_merged.vcf chr1.recode.vcf chr2.recode.vcf /
chr3.recode.vcf chr4.recode.vcf chr5.recode.vcf chr6.recode.vcf /
chr7.recode.vcf chr8.recode.vcf chr9.recode.vcf /
chr10.recode.vcf chr11.recode.vcf /
chr12.recode.vcf chr13.recode.vcf /
chr14.recode.vcf chr15.recode.vcf chr16.recode.vcf /
chr17.recode.vcf chr18.recode.vcf chr19.recode.vcf /
chr20.recode.vcf chr21.recode.vcf /
chr22.recode.vcf chrX.recode.vcf2、报错信息分析
是因为vcf的头文件没有contig这个描述。

正常的vcf文件应该包含contig。
如果你出现类似报错,可以检查一下是不是你的vcf文件缺少contig。
3、解决办法
vim varscan.indel.vcf使用vim对vcf文件进行编辑,进入文件后,按i键进行文本编辑,
如果对vim命令操作不熟悉,可以参考这篇文章
https://mp.csdn.net/mp_blog/creation/editor/137595595
进入编辑后,将下面的内容复制到头文件位置。
##contig=<ID=chr1,length=248956422>
###contig=<ID=chr10,length=133797422>
###contig=<ID=chr11,length=135086622>
###contig=<ID=chr12,length=133275309>
###contig=<ID=chr13,length=114364328>
###contig=<ID=chr14,length=107043718>
###contig=<ID=chr15,length=101991189>
###contig=<ID=chr16,length=90338345>
###contig=<ID=chr17,length=83257441>
###contig=<ID=chr18,length=80373285>
###contig=<ID=chr19,length=58617616>
###contig=<ID=chr2,length=242193529>
###contig=<ID=chr20,length=64444167>
###contig=<ID=chr21,length=46709983>
###contig=<ID=chr22,length=50818468>
###contig=<ID=chr3,length=198295559>
###contig=<ID=chr4,length=190214555>
###contig=<ID=chr5,length=181538259>
###contig=<ID=chr6,length=170805979>
###contig=<ID=chr7,length=159345973>
###contig=<ID=chr8,length=145138636>
###contig=<ID=chr9,length=138394717>
###contig=<ID=chrM,length=16569>
###contig=<ID=chrX,length=156040895>请注意,ID=chr1还是1,取决于你所用的参考基因组文件对染色体的描述。
插入完成后,保存并退出即可。