咱不来虚的,只分享干货,不谈枯燥的理论,只来通俗易懂的操作。先来看一张图:
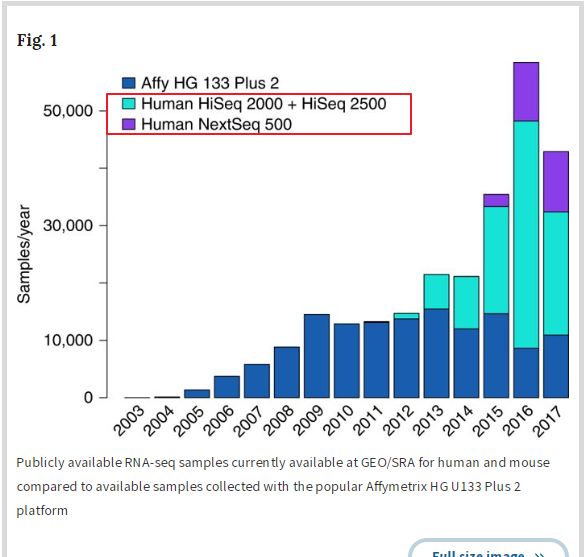
通过这张图展示的是 GEO数据库中的 RNA-seq数据与芯片数据积累随时间的变化,很显然测序数据从2015年开始就已经超过了芯片数据的累积 (生信宝典注:这里没有统计物种信息,芯片能应用的物种少,测序能应用的物种多。现在临床数据分析还是基于芯片的数据量更大一些,有兴趣一起易生信GEO/TCGA专题课程 - 挖掘公共数据,发表自己文章,同时适合GEO和测序数据)。大批量的数据产生固然是个好事,同时也带来了一个问题,公开的RNA-seq数据大多提供的是原始数据,这样就对数据的重新挖掘使用带来了很大困难。为啥嘞,数据量太大,临床医生,小实验室你确定做得了,就连测序数据从原始数据开始的分析都会遇到很多困难?
今天要介绍的神器呢叫做 ARCHS4,它的诞生呢就是为了解决这个问题,过程讲的比较复杂,简单讲就是西奈山医学院的Mayan实验室设计有效的算法把 GEO/SRA的 原始数据整合,分析,预处理成方便后续分析的矩阵格式。而且发了一篇Nature communication。就像 TCGA那样的数据库,之所以应用广泛,数据整理的格式就是原因之一呀。该数据库包括人和鼠的sample 187,946 , 其中人84,863,鼠103,083。接下来就看下具体这个神器有哪些功能吧:
数据下载功能Download
https://amp.pharm.mssm.edu/archs4/download.html (后台回复 转录组 获取链接)
可供下载的数据包括:
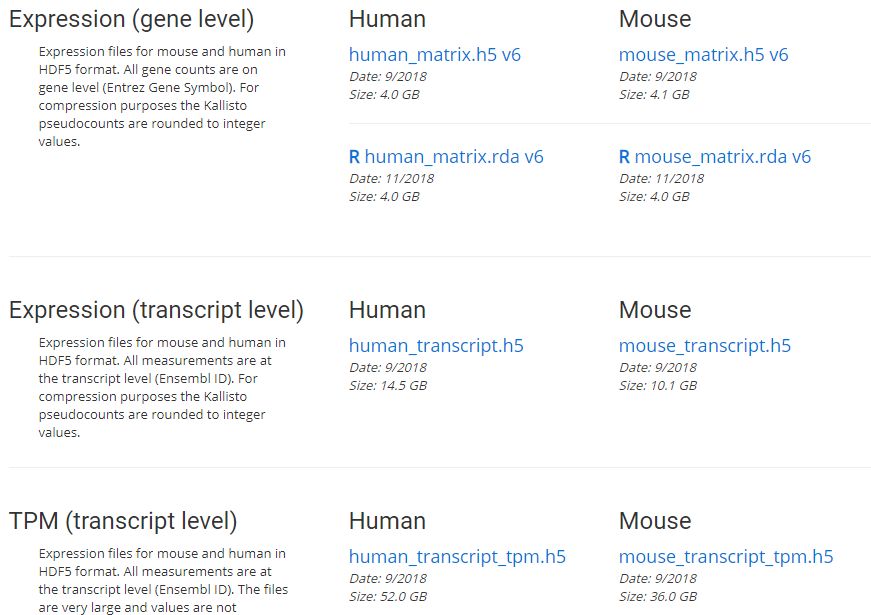
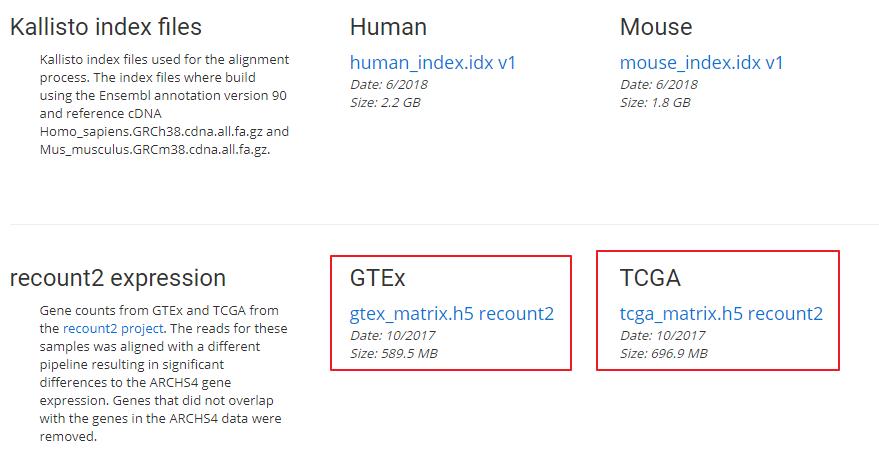
这里只列举了部分,甚至还包括了 GTEx/TCGA的数据,数据都整理为 H5格式,数据包括原始的 read count数据和 meta data信息,简单讲这些数据都整理成了方便后续分析的矩阵格式,可以这样全部下载。
当然也可以挑选自己感兴趣的下载,可以挑选自己感兴趣的组织,细胞系,也可以手动选择,基因集,Download部分会自动产生下载数据的 R代码,放到 Rstudio运行即可。
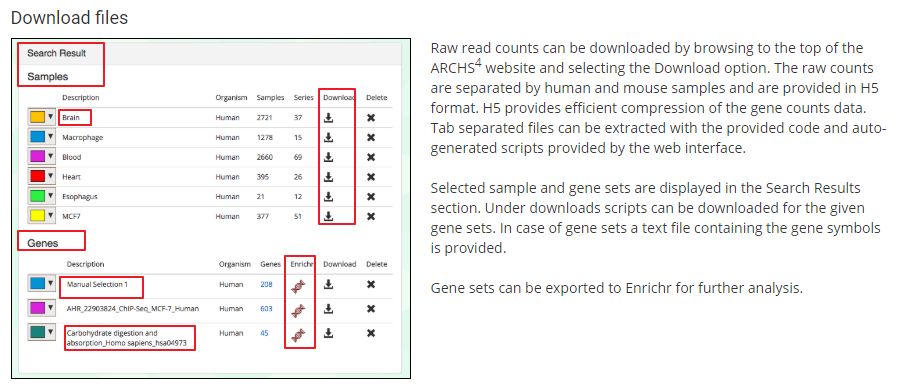
这里白介素同学,随便测试下载了一个代码,可以看看长啥样,有R基础的小伙伴应该更容易理解 (ggplot2高效实用指南 (可视化脚本、工具、套路、配色)):
大概就是这样,运行下就可以啦。
https://amp.pharm.mssm.edu/archs4/data.html#
此外数据下载后,就是做数据解析了,H5文件格式的解析,批次效应移除等,都有提供相应的代码 (DESeq2差异基因分析和批次效应移除):
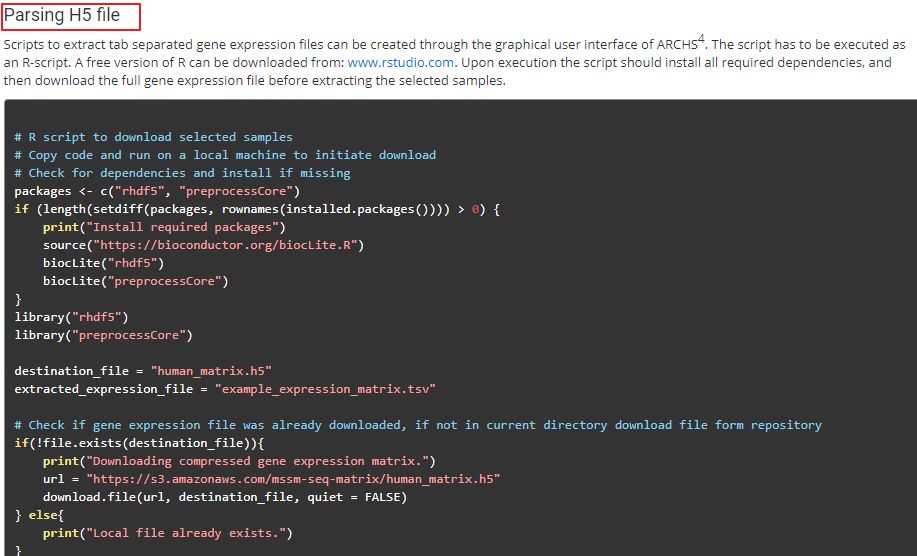
提供代码链接:https://amp.pharm.mssm.edu/archs4/help.html
查询功能
按 meta data信息查询,可以看下自己感兴趣的组织, 细胞系等的 (这个T-SNE聚类很有意思)。
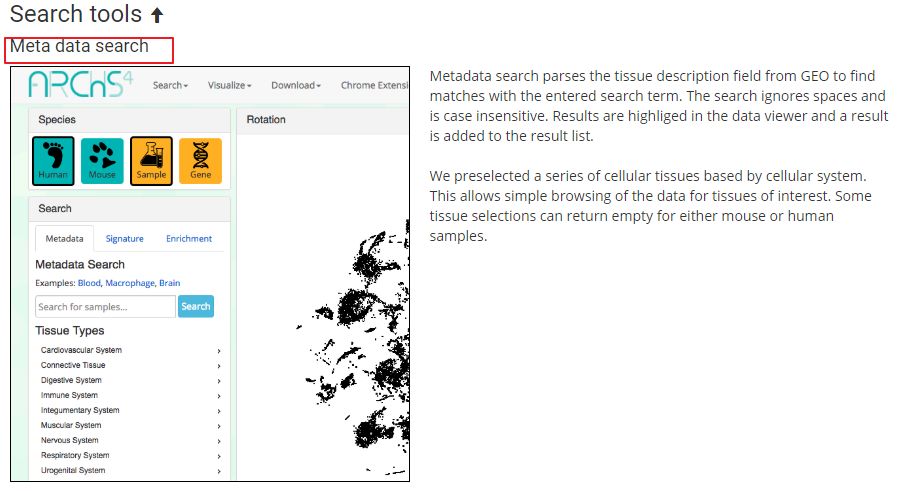
查找 signature,输入数据为上调和下调基因,寻找match这些基因的sample。

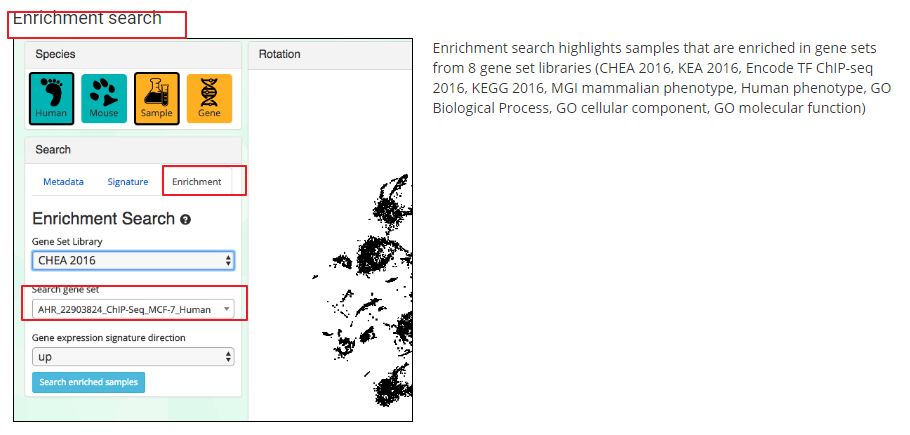
富集查询,找到富集某些基因集的sample然后下载,可以从8个基因集库中选择感兴趣的,比如KEGG库,GO库,其实这是一个反向的操作,与咱们通常的差异分析得到基因集进行富集不同,这是一种通过感兴趣的通路,基因集来找sample。
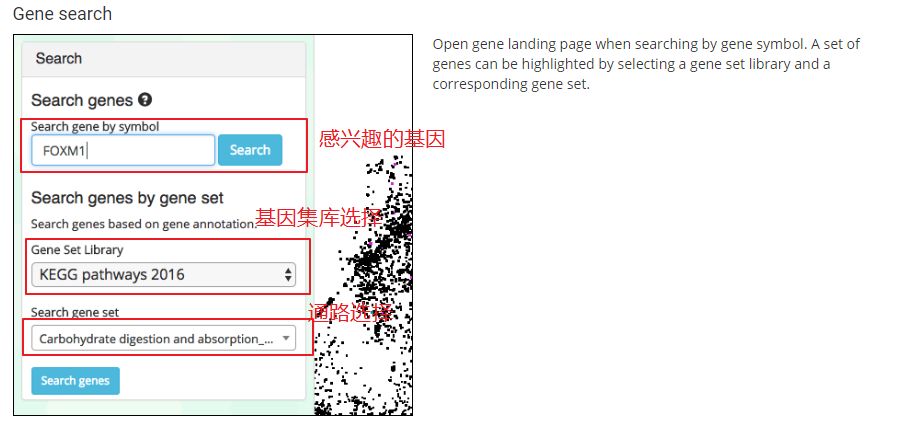
基因查询功能,遇到一个感兴趣的基因,这时候这个功能就派上用场啦,比如案例给出的 FOXM1基因
点击下就可以得到很多信息:
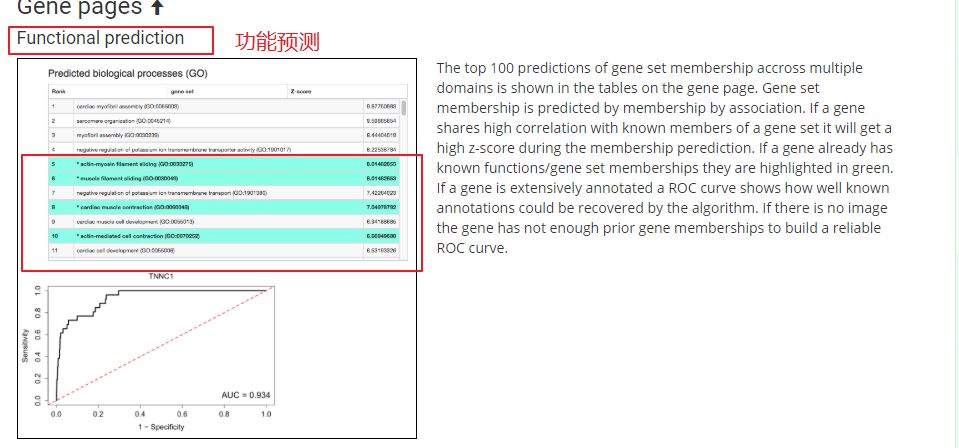

主要功能就是这些啦,提供处理过的数据下载和查询功能。然后就是了解下这个数据库的背景,文章在2018年4月发表在 Nature Communcations上。
内容就分享到这儿啦,白介素同学祝大家学习愉快!
附上网址:
https://amp.pharm.mssm.edu/archs4/index.html
参考资料:
https://www.nature.com/articles/s41467-018-03751-6