摘要 ✨
在全球化的医疗市场中,美国市场对于医疗器械制造商至关重要。本指南旨在为制造商提供全面的美国市场准入指导,确保产品合规并迅速进入市场。
监管机构
美国食品药品监督管理局(FDA)负责医疗器械的注册,确保其安全性、有效性和创新性。
主要法规
- 联邦食品、药品和化妆品法案(FD&C Act)
21 CFR(Title 21- Code of Federal Regulations Parts 1-58, 800-1299)
参考法规
- 国际医疗器械监管机构论坛(IMDRF)指南
美国国家标准协会(ANSI)标准
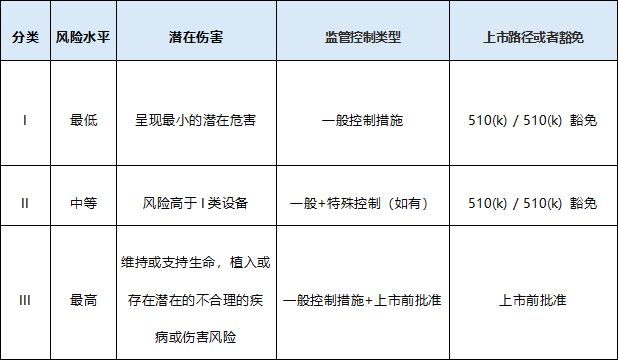
医疗器械风险分类
本分类确保不同风险等级的医疗器械得到适当的监管:
符合性评估路径
FDA提供多种评估路径以适应不同医疗器械的需求:
- 510(k):适用于大多数医疗器械的上市前通知。
- PMA(Premarket Approval):适用于高风险医疗器械的上市前许可。
De Novo 分类:适用于新类型的低到中等风险医疗器械。
FDA官方费用(2024年)
以下是不同申请类型的标准收费和小企业资质收费:

审批时间
注册时间根据产品风险等级分类的不同,从几周到几个月不等。FDA的目标是在90天内做出510K审批决定。
申请资料
- 510(k)提交:eSTAR及其附件。
PMA提交:eCopy,包括更为详尽的临床数据和研究报告。
注册申请流程
通过FDA的电子提交系统在线提交,简化注册过程。注册证有效期
审批后,需交年费、进行产品列示和企业注册以维持正常销售。
语言要求
所有提交的文件和说明书必须使用英文。注册资格
海外制造商需指派一名美国代理人,该代理人必须是美国公民或合法实体,其角色主要是帮助FDA与海外制造商之间的沟通、协助FDA安排监查等。证书转让
FDA容许某器械的510(k)证书从一个公司转移到另一个公司。医疗器械注册流程概述
确定产品是否属于医疗器械及风险分类
指定美国代理人
准备注册文件
提交注册文件至FDA
获取医疗器械注册证书
- 合法进入美国市场销售