目录
1,单细胞早期、晚期和转移性 LUAD 的细胞动力学变化
2,细胞代谢重编程介导的LUAD驱动恶性转移的异质性
3,模型构建 S-MMR评分管线构建
4,S-MMR 模型的预后评估
5, 还开发了S-MMR 评分网络工具
6,S-MMR 评分重塑了 LUAD 中的免疫浸润模式
7,S-MMR评分预测免疫治疗疗效的能力
8,靶点和药物筛选
9,解剖 S-MMR 评分为 3 的恶性细胞
10,泛癌分析
最近的研究越来越多地揭示了代谢重编程与肿瘤进展之间的联系。然而,代谢重编程对肺腺癌 (LUAD) 患者间异质性和预后的具体影响仍需进一步探索。在这里,我们根据恶性和代谢基因集引入了一个细胞层次结构框架,称为恶性和代谢重编程(MMR),以重新分析178,739个单细胞参考图谱。
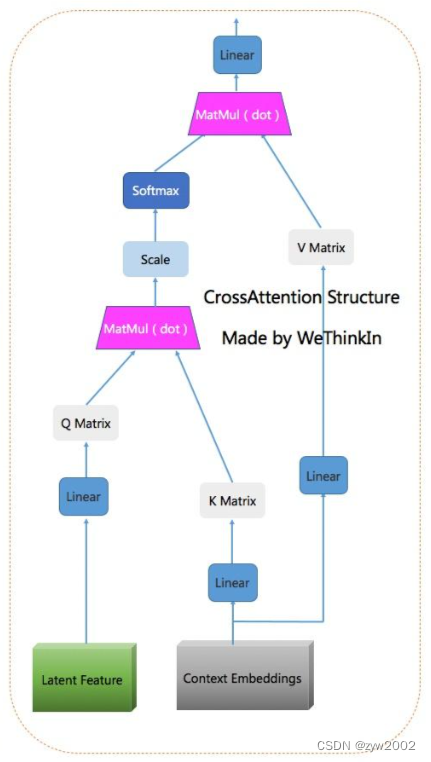
亮点:大工作量,支持向量机、随机森林以及决策树模型等多机器学习框架(比单独机器学习模型和算法具有更好的稳健性和精确性)。该研究根据 LUAD scRNA-seq 图谱定义了一组与 LUAD 肿瘤发生和细胞代谢重编程相关的基因,命名为“MMR”。采用Cox回归、随机生存森林(RSF)、CoxBoost、支持向量机(SVM)和梯度提升机(GBM)等机器学习方法,明确了MMR与LUAD预后的关系。我们引入了一种创新的集成学习管道,即三阶段 MMR (3 S-MMR),并通过遗传算法进行增强。该框架分别在特征工程和模型开发中使用双训练集,从而降低了严重过拟合的风险。研究涉及了单细胞、空间代谢组学多组学研究。
1,单细胞早期、晚期和转移性 LUAD 的细胞动力学变化

在解开LUAD细胞层次结构的初始阶段,我们重新分析了178,739个scRNA-seq细胞,覆盖48个样本,包括Nln、nLung、tLung、PE、mLN和tL/B组织,以及根据经典标记基因对T、B、NK、上皮、巨噬细胞、单核细胞、成纤维细胞、MDC、肥大、血浆、内皮和PDC进行明显分类的细胞。
早期、晚期和转移性 LUAD 的细胞动力学变化。(A)样品的细胞分布无显著的批次效应。(B) 来自所有 scRNA-seq 样品的细胞的 t-SNE 图谱,通过细胞类型注释着色。(C) 显示每种细胞类型的代表性标记基因的点图。(D) 每种细胞类型中来自每种来源组织的比例,如图所示。(E) 折线图显示通过 Ro/e 评分估计的每种细胞类型的组织流行率。(F) 分级热图显示来自每个来源组织的上皮细胞的 CNV。正常肺源性上皮细胞用作对照参考。红色:增益;蓝色:损失。(G) 推断 CNV 的 K 均值聚类以获得癌细胞。(H) 显示5个K-means聚类CNV分数差异的小提琴图。(簇1被指定为正常上皮细胞,而其余细胞被归类为恶性细胞。)
2,细胞代谢重编程介导的LUAD驱动恶性转移的异质性

由细胞代谢重编程介导的 LUAD 驱动的恶性转移之间的异质性。(A) 正常细胞和恶性细胞之间 GSVA 对每个细胞评分的标志性基因集通路活性的差异。(B)来自每个来源组织的恶性细胞的代谢途径活动。统计上不显著的值(随机排列余P > 0.05)显示为空白。(C) 基于恶性细胞和正常细胞之间差异表达基因的 Wilcoxon 秩和检验结果的百分比差异(Delta 表示细胞百分比)和对数倍数变化。(D) 显示 1290 个 MMR 基因交叉分析的 UpSet 图。(E) 1290 MMR基因的DO富集分析。(女、女)小提琴图 (F) 和气泡图 (G) 显示使用 AUCell、UCell、singscore、ssGSEA、AddModulescore 和 Scoreing(其他算法的分数之和)评分的每种细胞类型的 MMR 基因集的富集分数。(H) 使用 AUCell、UCell、singscore、ssGSEA、AddModulescore 和 Scoring 评分显示各来源组织的 MMR 基因集富集分数动态变化的小提琴图
3,模型构建 S-MMR评分管线构建

(A) 3 S-MMR 评分的工作流程。(B) 25个LASSO驱动基因对的基因对信息和危害比。(C) 47名基础学习者的C指数和标准
4,S-MMR 模型的预后评估

5, 还开发了S-MMR 评分网络工具

6,S-MMR 评分重塑了 LUAD 中的免疫浸润模式

3 S-MMR 评分重塑了 LUAD 中的免疫浸润模式。(A) 高 3 S-MMR 评分组和低 3 S-MMR 评分组之间癌症免疫周期各个步骤的差异。(B) 3 S-MMR 评分 (riskScore) 与基质、免疫和 ESTIMATE 评分之间的相关性。(C) 3 S-MMR 评分与癌症免疫周期步骤之间的相关性(左)。3 个 S-MMR 评分与已发表的免疫细胞特征的富集评分之间的相关性(右)。(D) 3 S-MMR 评分与 6 种 TIIC(CD8 + T 细胞、CD4 + T 细胞、NK 细胞、巨噬细胞、Th1 细胞和树突状细胞)浸润水平之间的相关性,采用 6 种独立算法计算。(E) 表示高 3 和低 3 S-MMR 评分组之间病理 HE 染色变化的图像(TCGA 数据库)。(F)从左到右:mRNA表达(中位归一化表达水平);表达与甲基化(基因表达与 DNA 甲基化 β 值相关);扩增频率(在特定亚型中扩增 IM 的样本分数与所有样品中的扩增分数之间的差异);以及高 3 和低 3 S-MMR 评分组对 75 个 IM 基因的缺失频率(作为扩增)。缩写:*P < 0.05;**P < 0.01;P < 0.001。
7,S-MMR评分预测免疫治疗疗效的能力

S-MMR 评分预测免疫治疗效果的能力。(A-F)TIDE (A)、功能障碍 (B)、排除 (C)、CD8 (D) MDSC (E) 和 Merck18 (F) 评分的小提琴图。(G) 子图算法预测高低 3 个 S-MMR 评分组对 CTLA4 和 PD-1 抑制剂的反应。(H) 高低 3 S-MMR 评分组患者之间免疫检查点曲线的相对表达水平的箱线图。(I-N)GSE126044 (I-J)、GSE35640 (K-L) 和 GSE78220 (M-N) 队列中免疫治疗反应者和非反应者之间 3 个 S-MMR 评分的差异。(O-P)T-SNE 降低映射了 SD 和 PR 患者 (O) 的细胞分布,以及 GSE207422 数据集中 3 个 S-MMR 评分 (P) 的分布。(Q) GSE207422数据集中 SD 和 PR 患者之间 3 个 S-MMR 评分的小提琴图。(R) 通过 R 估计高低 3 个 S-MMR 组的组织偏好O/E在GSE207422数据集中。(S-T)T-SNE 降低映射了 SD 和 PR 患者 (S) 细胞的分布,以及 GSE145281 数据集中 3 个 S-MMR 评分 (T) 的分布。(U) GSE145281数据集中 SD 和 PR 患者之间 3 个 S-MMR 评分的小提琴图。(V) 高低 3 个 S-MMR 组的组织偏好通过 R 估计O/E在GSE145281数据集中。缩写:*P < 0.05;**P < 0.01;P < 0.001
8,靶点和药物筛选
首先,我们进行了 Spearman 相关性分析,以探索 TCGA-LUAD 队列中 3 个 S-MMR 评分与潜在药物靶点表达水平之间的关联。由此,我们确定了一组与评分呈正相关的共享基因,将这些基因指定为 3 S-MMR 评分的相关靶标。随后,通过使用肺癌细胞系对 CERES 评分和 3 S-MMR 评分进行 Spearman 相关性分析,我们继续确定 54 个依赖于不良预后的靶点。

9,解剖 S-MMR 评分为 3 的恶性细胞

解剖 S-MMR 评分高 3 的恶性细胞。(A)Monocle2推断的恶性细胞的发展轨迹。3 S-MMR 评分高的恶性细胞主要位于分化根部,3 S-MMR 评分低的恶性细胞主要位于中点和终点状态。(B)恶性细胞中3个S-MMR评分相关基因沿假时间的热图。(C) 热图显示了高 3 S-MMR 评分恶性细胞和低 3 S-MMR 评分恶性细胞之间不同 TFs 激活的热图。(D、E)TFs 在恶性细胞高 (D) 和低 3 S-MMR (E) 评分之间的最高活性。RSS 表示调节子特异性评分。(女、女)所有细胞类型的细胞聊天分析。显示了相互作用的数量和相互作用强度。(H,I)显示 SPP1 信号通路推断的细胞间通信网络的分层图。(J) HE染色显示stRNA样品的组织学不同区域。黄色:癌症区域。(K) 3 S-MMR评分强度的空间图。(L)利用RCTD算法识别空间图中不同细胞类型的分布。
10,泛癌分析
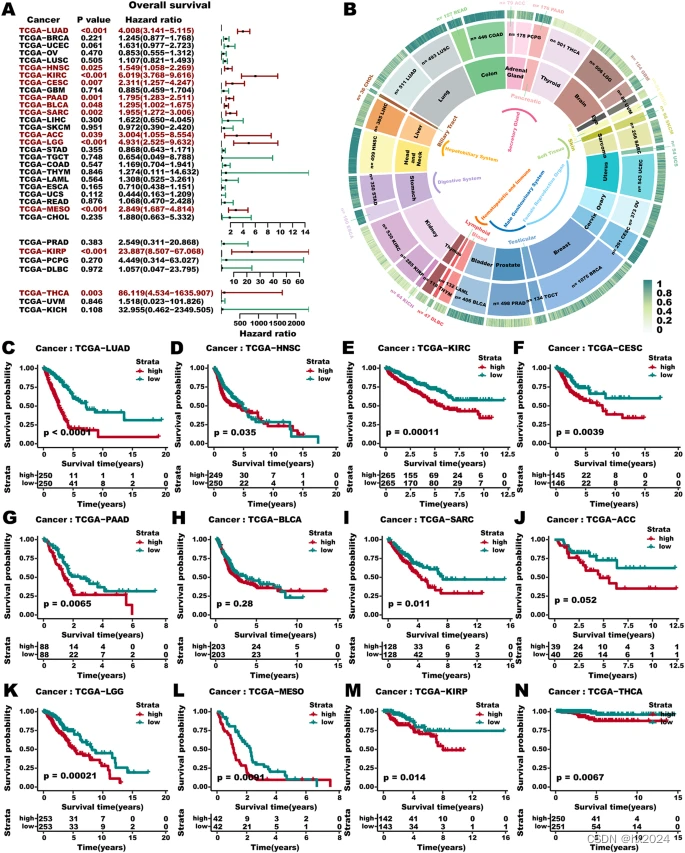
(A) 33 种癌症类型中 3 个 S-MMR 评分的 Cox 回归分析。红色表示 P < 0.05 显著性结果。(B) 个别癌症类型的平均 3 S-MMR 评分。组织类型、癌症类型和平均 3 S-MMR 评分从内圈到外圈显示。(C-N)在 12 种癌症中,3 个 S-MMR 评分的 Kaplan-Meier 生存曲线显著(对数秩检验)
参考文献:Architecting the metabolic reprogramming survival risk framework in LUAD through single-cell landscape analysis: three-stage ensemble learning with genetic algorithm optimization